芯片作为一个成熟的技术平台,几十年来被广泛用于基因表达谱的检测。近年来,二代测序技术(Next Generation Sequencing , NGS)通过量化整个转录组上的转录本reads密度来检测基因表达水平,被越来越多的应用于基因表达研究。长链非编码RNA(Long non-coding RNAs , LncRNAs)是长度超过200nt的非蛋白编码转录本。LncRNAs在正常的生理过程和疾病中发挥重要功能,已成为科学研究热点。对于LncRNAs基因表达谱检测,芯片技术比RNA-seq有许多重要且不可替代的优势,仍然是LncRNAs表达谱检测的优质平台(表1)。
LncRNAs比蛋白编码RNAs表达水平低
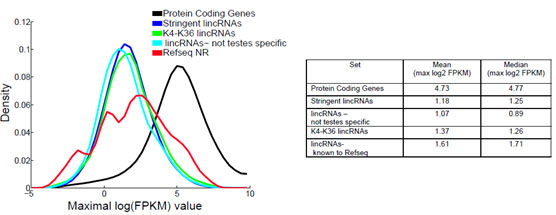
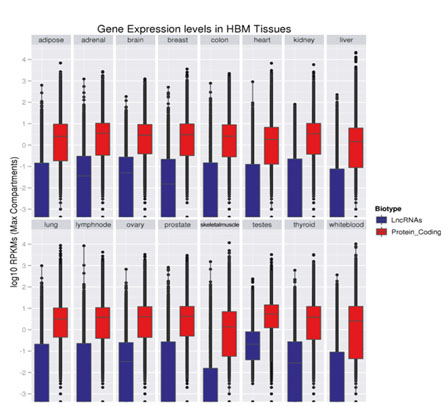
LncRNAs的一个普遍特征是表达水平很低[1-5]。根据FPKM定量(每1百万个匹配上的fragments中,匹配到外显子的每1千个碱基上的fragments个数),LncRNAs的平均表达水平约为蛋白编码基因的1/10(图1和图2)[4-7]。
|
图1. RNA在所有组织中的最高表达水平分布曲线。log2FPKMs值经由Cufflinks评估,蓝线表示严谨型lincRNAs,绿线表示含有K4-K36结构特征的严谨型lincRNAs,浅蓝色线表示非睾丸特异性的严谨型lincRNAs,红线表示RefSeq NR中已知lincRNAs,黑线表示蛋白编码基因。右表为最高表达水平的平均值和中值[4]。蛋白编码基因与lncRNAs的表达差异倍数表示为2Dlog2(FPKM)。 |
|
| 图2. LncRNAs(蓝色)和蛋白编码转录本(红色)在人体组织中的表达分布[7]。纵轴上的表达水平以log10RPKM表示。 |
RNA-seq对于低丰度转录本的定量不可靠
对于低丰度的RNA,由于测序深度有限造成的泊松抽样误差是RNA-Seq误差的主要来源。因此,低表达的转录本不能被RNA-seq可靠检测,需要进行富集检测[8]。
在一个包含331M 50bp reads的RNA-Seq数据库(三次技术重复)中分析发现基因表达水平与测量精度之间的联系[9]。如图3所示,基因表达水平越高,定量越准确;反之,基因表达水平越低,定量的相对误差越高。由于LncRNAs的表达水平远低于蛋白编码基因,绝大部分低丰度的lncRNAs无法通过RNA-seq准确定量(相对误差高于20%)。
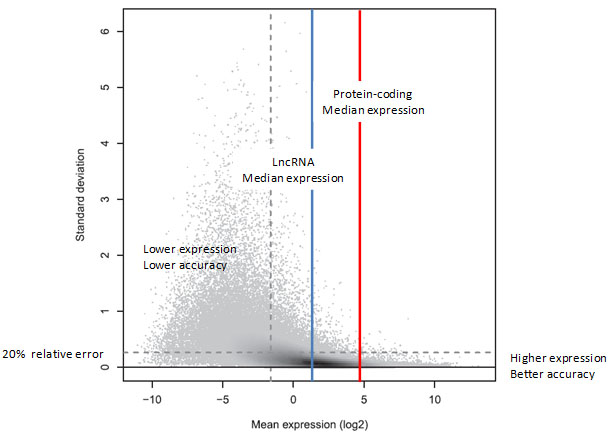 |
图3. 标准偏差与表达水平的关系。Y轴为三次技术重复的标准偏差,每个灰色点代表一个转录本。在阴影区,灰度代表密度,灰度越深,密度越大。如图中所示,转录本平均表达水平越低,标准偏差越高;表达水平越高,标准偏差越低,定量越可靠,相对误差低于20%。在RNA-Seq中,只有41%的转录本能够被准确定量(水平虚线以下);而对于高表达的转录本(垂直虚线右侧),其中高达84%的转录本的定量是可靠的。这可以从垂直虚线右侧而不是左侧的高密度区落在了水平虚线以下看出[9]。 |
增加测序深度不能提高低丰度转录本的检测准确度
增加RNA-Seq测序深度可以提高表达谱的检测准确度。通常,100M reads对于大部分基因和转录本的检测是足够的,但如果要对多数(72%)基因的表达水平进行准确定量则需要500M reads[10]。然而,在对不同丰度的PHB、CD74和BRD4转录本亚型的研究中发现,提高测序深度能够明显提高高丰度转录本的检测准确度,但是并不能提高低丰度转录本的检测准确度 [10]。
总的来说,无限制的增加测序深度并不能无限的提高低丰度转录本表达水平的检测准确度。由测序深度增加而提高的准确度将逐渐趋于饱和(图4)[9]。在RNA-Seq数据中,仅7%的高丰度转录本占总测序reads数的比例高达75%。增加的数据量绝大部分浪费在少量高丰度的转录本上,例如管家基因。这意味着低丰度lncRNA的表达水平不仅在通常的测序深度下不能够准确检测,即使尽可能增加测序深度也不能够实现对低丰度lncRNAs的准确检测。此外,随着测序深度的增加RNA样品中加工不完全的RNA检出率提高,从而引起LncRNA检测准确性的下降。
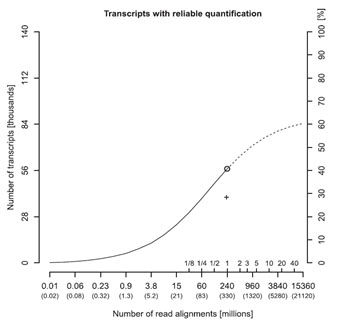 |
图4. 定量可靠(误差低于20%)的转录本数目与测序深度的关系。X轴的带括号数字表示总测序reads数。额外的内部刻度线表示值得测序分析的测序运行数。右Y轴为所有已知的可被可靠定量的转录本的比例。小圆圈表示在完整的测序运行中的reads数(331M reads)。外推的S型曲线表示RNA-Seq能可靠检测的转录本最多只能到60%,即使reads数达到10bn。 |
RNA-Seq不能精确定量LncRNA与RNA-Seq数据分析不成熟密切相关
迄今为止,RNA-Seq数据分方法在外显子检测及RNA定量等过程中存在诸多误差。例如:1)对LncRNA外显子序列的识别效率低。蛋白编码基因的外显子可以通过参考基因组上的翻译起始位点,终止位点,以及剪切供体和受体位点直接识别,而LncRNA无法通过这些特征位点直接识别。此外,LncRNA的表达水平比蛋白编码基因低,即使在相同的覆盖深度下,检测的灵敏度也低于蛋白编码基因3)对LncRNA的组装精度很低(图5),而且无法通过增加测序深度提高组装精度。这是由于RNA样品中含有加工不完全的RNA或是转录噪音,随着测序深度的增加它们被测出来的频率越高,因此随着测序深度的增加,组装精度不断下降。4)定量不准确。通过比较RNA-Seq与NanoString的结果发现:RNA-Seq的定量结果与NanoString之间相关性很低(R: 0.34-0.68),而且很多被NanoString检测到的转录本通过RNA-Seq无法检测到。
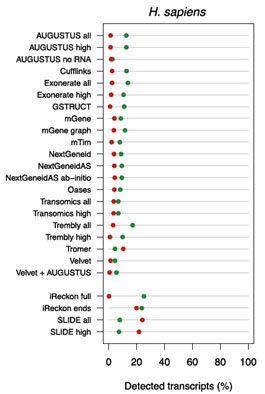 |
图5. 通过RNA-Seq数据进行LncRNA组装的精度很低。图中横轴代表正确组装的转录本占已报道的人类转录本的比例,绿点代表蛋白编码转录本,红点代表非编码转录本(LncRNA)。能被正确组装的蛋白编码转录本不到已知转录本的30%,而能被正确组装的LncRNA比例更低,只有不到20%。 |
芯片比RNA-Seq更适合低丰度lncRNA 表达谱的检测
芯片的原理是通过与序列特异性探针的杂交识别RNA。对于特定的基因,即使杂交体系中存在高丰度的无关序列也不会影响该基因的杂交结果,因而对于低丰度表达谱的检测几乎无影响。而RNA-Seq的数据中,大部分测序reads被表达丰度很高的RNA如管家基因占据,从而导致低丰度的RNA只有很低的覆盖深度。低覆盖深度意味着低灵敏度和低可靠性。因此,芯片更适合低丰度RNA的检测。例如,芯片一般能够检测到7000~12,000个 LncRNAs,而RNA-Seq 多达120M reads也只能够检测到1000~4000个 LncRNAs [11]。
虽然RNA-seq可以同时检测已知和未知的序列,但随着数十年的人类表达序列检测的数据累积,RNA-seq能发现的新转录谱越来越少[10]。更多时候,RNA-seq文库构建时产生的人为序列和不成熟的RNA会被误认作新的转录本。随着高级的芯片设计,转录本亚型可以被芯片上针对外显子连接点的特异性探针所区分。涵盖绝大多数已知lncRNA并且能够特异性检测转录本亚型的芯片,由于具有比RNA-Seq更高的灵敏度,仍然是研究者的优先选择。
表1. 芯片和RNA-Seq对lncRNAs表达谱检测的比较
芯片 |
RNA-Seq |
不受高丰度RNAs的干扰,序列特异性的芯片探针可以高效的识别低丰度的LncRNA。 |
基因表达水平越低,检测效率越低。低丰度RNAs的测序深度会被高丰度RNAs降低,导致低丰度RNAs的检测不可靠。 |
对于低丰度RNAs,如lncRNAs,有更高的灵敏度[12]。芯片通常可以检测到7000~12000个lncRNAs。 |
对于低丰度RNAs,灵敏度很低。多达120M的reads只能检测到1000~4000个lncRNAs[11]。 |
实验流程简单成熟,在aRNA的合成/标记过程中没有序列偏好性 |
大部分测序技术在建库过程中对样品进行了PCR扩增,而PCR对高GC含量区域的扩增效率不高,从而导致了后续的测序偏好性。 |
数据分析流程简单,方法成熟,对LncRNA的分析误差小。 |
数据分析流程繁琐,方法不成熟,在LncRNA的数据匹配,组装,定量等过程中存在诸多误差。例如:1)RNA样品的来源与参考基因组的来源不同,在匹配过程中往往存在差异。2)对LncRNA外显子序列的识别效率低。3)对LncRNA的组装精度很低。4)定量不准确。 |
芯片的操作过程,例如样品标记和芯片杂交可以同时应用于大量样本。对于大量样本的研究和含有很多数据点的科研项目,更高的样本同时处理能力是至关重要的 [12]。 |
RNA-Seq一次只能对一个或几个样品进行测序,延长了时间。 |
References
1. Kampa, D., et al., Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res, 2004. 14(3): p. 331-42.
2. Cawley, S., et al., Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell, 2004. 116(4): p. 499-509.
3. Ravasi, T., et al., Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res, 2006. 16(1): p. 11-9.
4. Cabili, M.N., et al., Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev, 2011. 25(18): p. 1915-27.
5. Guttman, M., et al., Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol, 2010. 28(5): p. 503-10.
6. Yan, L., et al., Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol, 2013.
7. Derrien, T., et al., The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res, 2012. 22(9): p. 1775-89.
8. Jiang, L., et al., Synthetic spike-in standards for RNA-seq experiments. Genome Res, 2011. 21(9): p. 1543-51.
9. Labaj, P.P., et al., Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics, 2011. 27(13): p. i383-91.
10. Toung, J.M., et al., RNA-sequence analysis of human B-cells. Genome Res, 2011. 21(6): p. 991-8.
11. Kretz, M., et al., Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes Dev, 2012. 26(4): p. 338-43.
12. Xu, W., et al., Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci U S A, 2011. 108(9): p. 3707-12.
13. The_ENCODE_Consortium, Standards, Guidelines and Best Practices for RNA-Seq. http://encodeproject.org/ENCODE/protocols/dataStandards/ENCODE_RNAseq_Standards_V1.0.pdf, 2011. |