
-

����ͨ��
����ץס�����Ƽ�
����������
Science:ͨ��ȫ����������ʾDNMͻ����ǿ��֢�е�Ⱦɫ�������й�
�����壺 �� �� С �� ʱ�䣺2022��01��14�� ��Դ��AAAS
�༭�Ƽ���
�������о����ڶ�ʶ�������������Ʊ���ij�������DZ�ڵĸ߶��������Ŵ������������������Ȥ���״�ͨ��WGS����ȫ������ DNM ���������˻���ǿ��֢�ļ��塣�о����������2022��1��12�յ�Science��־�ϡ�
ǿ��֢��OCD����һ�����Խ���֢��������Ҫ���Ŵ������㷺δ�����ֵIJ��������ǿ��֢�Ĵ�ͷͻ�� (DNM) ������������о���ǿ�˺������쵼�·��յļ��衣��������֪�����Ƕ� 53 ����ǿ��֢Ӱ��ĺ�������Ӽ�������˵�һ��ȫ����������Ե������к����ģ�DNM������Ͳ���/ȱʧ�����ǹ۲쵽������ê��Ⱦɫ�ʻ�������P = 0.0015�����鵰�ױ��Ƶ�ʸߵ�������P= 0.0001)��Ӱ���������ͻ����������Է��������в���Ⱦɫ�����εĻ���Ĺ�����ģ�������Ÿ�������genes- SETD5��KDM3B��ASXL3��FBL -hadǿ������֤�ݻ��ܺ����ں�ת¼�ı����Ŵ����أ�����ζ��һ����Ҫ��OCD���ջ��ơ����ǵ����ݱ����˲�ͬ��ȫ������ DNM����ǿ����Ⱦɫ�������� OCD ����ѧ�е�Ӱ�졣
����
ǿ��֢��OCD����һ�����ص������ϰ��������������ظ��ģ�ǿ���Ե��뷨����/��ǿ���˶�����ʽ����Լ1-3%���������������1 - 3����ǿ��֢�����ͨ�����ڹ۲쵽����Ϊ��֢״����ʱ������������ǻ��ڶԼ������������ѧ���⡣�������谭���ڿ�������ȷ����Ϻ��õ������Ը��ƻ���Ԥ����Ľ�չ�����в�ѧ�о��Ѿ�֤���Ŵ����ض�ǿ��֢�������ҪӰ�졣����ͬ��˫��������˫��̥�о���һ���ʣ�OCD ���Ŵ���ԼΪ 50%��4�������ܴ�ͳ�Ŵ�ѧ�о��Ѿ�ͻ����һЩ�ٶ���Ⱦɫ������ ( 5 )������δȷ�� OCD ���²����� ( 6 )��
�����ȫ����������о� (GWAS) ( 7 , 8 ) �Ѿ�ȷ������ OCD ��صĵ��������̬�� (SNP) �ڹȰ����źŴ�����ͻ�������е����á�Ȼ�������� SNP �� OCD �Ŵ�������Ϊ 0.22 ( 7 )��������Լ������յ�ʣ��ײ���������صij��� SNP ( 9�����������в�ѧ�о���ǿ��֢������Ŵ����� GWAS ���Ƶ�����������֮�������ì�ܣ�����ν�ġ�ȱʧ�Ŵ��ԡ��������ˡ���ͷ��ʽ���ļ��裬�������DZ�ڵ�����ǿ��֢��չ�����еĺ������졣�����Cappi����������ǿ��֢���������������ȫ����������� (WES) �о���( 10 , 11 ) �Ѿ���ʾ�������ŷ���֤��֤��ͻ�� (DNM) �� OCD �е����ã����һ��֤���ˣ�DNM�������ھ������Ŵ��ṹ�о�����Ҫ���õļ��� ( 12 )������ǿ��֢���������о� ( 10 ,11 )��������������ֱ� = 20 �� 184������Լ 22% �� OCD ����Я���ɹ��Ƽ����������ƻ��Ա��� DNM��ͬʱ������ǿ������ǿ��֢�IJ���ѧ�о���DZ�����õ���������CHD8��SCUBE1��
Ȼ�������������ǿ��֢���ԵĴ��ģ�о���������ͨ��������� ( 13 ) ������������� ( 10 , 11 )���������չ����ڻ�����ĵ����ʱ�������������䡢�DZ��� RNA (ncRNA) �ʹ�ṹ���� (SV) ���� ( 14 , 15 ) �����Եĸ������²�����Ҳ����֪�ģ������ȫ��������� (WGS) ����ͻ�����ʹ�С��WES�����Ա���Ϊ���о�ǿ��֢����ѡ������ƽ̨�����⣬WGS �����������ϰ��о��е�Ӧ�ã������Ա�֢��ϵ (ASD) ( 14 �C16 ) �������ϰ� (ID) ( 17 ) �Ѿ�չʾ�������˽�������ģ�� DNM �����帺������Լ����Ĺ�����������ױȵ�������
��ˣ���֮ǰ�� OCD �����������о�֮������Ӧ�� WGS ��������Ϣѧ�������о�һ�� OCD ���������࣬��ʶ��������ģ�Ĵ�ͷ���죬��������������� (SNV)��С�����ȱʧ (indels ) �� SV���� 53 �����Ӽ�ͥ�У����Ƿ�����ǿ������֤�ݱ���������������õĻ���ͻ�䷢���ʺܸߡ����ǻ��۲쵽 DNM ���ȷ����ڲ����ܻ����Ӱ��Ⱦɫ�����εĻ����ϡ����⣬���Ǽ��������ָ����Ŷȵ�Ⱦɫ������������SETD5������ SET �Ľṹ�� 5����KDM3B��������ȥ����ø 3B����ASXL3��ת¼�������� 3)������Ϊ���ڶ���֤�ݵ�ǿ��֢��ѡ���ջ���������ǵ��ۺϷ������֣���ǿ��֢��ͼ�����ϰ� (TD) DNM ( 18 �C 20 )Ӱ��Ļ����������Ƶ�����ѧ���ܣ�����Ӱ�첻ͬ�Ĵ��������ϸ�����ͱ���ģʽ������һ���̶��Ͻ���������֮�临�ӵĹ�����ϵ�����������ϰ���
���Ƕ����� 53 ����ͥ�� 160 �� DNA ���������� WGS������ 54 ��ǿ��֢������δ�����ؾ���Ӱ��ĸ�ĸ�����Ϻͷ����Լ��� S1�����������ƺ����� 51 ���������һ��������� 53 ����֤���Լ�����δ��Ӱ��ĸ�ĸ���������ķ����У�����һ���������ͥ���Ƴ�������������Ϊ 157 �ˣ���ƽ�����ԣ�89.1% �Ļ����������ڸ�����汻 20 �λ����������ǡ�ƽ���������Ϊ 29.92����ͼ S1������ 53 �����Ӽ�ͥ�У��������ȷ���� 4143 ���µ� SNV �Ͷ̲���ȱʧ������~96% �����ʽ��� Sanger ������֤�μ����Ϻͷ����Լ��� S2�������DZ����� 4062 �� DNM �б����� S2����ͻ����Ϊ 1.34 �� 10 -8��95% �������䣺1.26 �� 10 -8�� 1.41 �� 10 -8��ÿ������ԡ�ÿ����֤�ߵ� DNM �����ڻ������д� 51 �� 117 ���ȣ����������д��㵽�ĸ�����35 ����֤��Я��һ������������ DNM����ÿ��������DNMS����0.93��SNV��0.13��������IJ���ȱʧ��62.5���ڸ�SNV���Լ�14.7���������飬�������ھ�����������ͥΪ������WES��WGS�о��������ȱʧ��14 - 16 )��
�����������Ǽ�����ȫ������һ��ͻ�����͵����ͻ���ʣ��ض����͵� DNM ���������о��� DNM ���������������ӡ��ں��ӡ��Ƿ����� (UTR)��ncRNA �ͻ���䡣�Ľ��������ǰ��ASD��WGS�о�������14����OCD��֤�ߺ�Simons Simplex���ϼ���SSC���ؼ���֮��IJ���15�ڴ�������ͣ�û�дﵽ�����˷DZ���RNA�Ķ��ؼ���У����ͳ��ѧ���壨P = 0.02)�����⣬���ǽ��ƻ��� DNM �����Ϊ���´�ͷ����ɥʧ��LoF���������ȱʧ��ֹͣ��������λ��ͻ�䣩���²�����[�� SIFT ( 21 ) �� PolyPhen-2 (22 ) �����ƻ���Ӱ�죬�Լ����ע�������ľ� (CADD) ( 23 ) �� 20]�� PHRED ���������Ȼ������ͨ��������ͬ��ͻ���ʵ����������ͻ���������� OCD �� SSC ���յ��ƻ���ͻ���ʣ� 15������Ϊʹ��ͬ��ͻ����Ϊ�ڲ�����Ӧ���ܹ��ֿ��ɱȽ������DZ��αӰ�����Բ�ͬ�о������ݡ����ǹ۲쵽���Ÿ��ߵ�ͻ���ʵ��� OCD �е� DNM [ P = 0.02�����Ʊ� (OR) = 2.32]�����⣬������-����ऺ�����GC ��������Wilcoxon ����P = 0.1������ȣ�Wilcoxon ����P = 0.9���ڲ����Ͷ����е� DNM ����֮�䣬�����۲쵽�� OCD �ƻ���ͻ���ʵ������Բ�����ƫ������ģ����� GC ��������ȡ�
���ţ������о���OCDͻ���Ƿ��ڹ��ܻ�������������Ƶ����ͨ���������Զ����ͬ������Դ�Ļ�����ע�ͣ�����PsychEncode��24����ENSEMBL��25����������ͼ���ۻ����飨26������У����β��Ժ�����û�й۲쵽 DNM ֱ�����������ӡ���ǿ�ӻ���зDZ�������ȫ�������ȫ�ָ�������Ҳ������֮ǰ�Ծ������֢ (SCZ) ������Ĵ��� WGS �о� ( 14 )��Ȼ�������ǹ۲쵽���ǵ� OCD ͻ����������ê����Ⱦɫ�ʻ������Ÿ���������PУ��< 0.001 �� OR = 1.23��ͼ 1A)������������������õ�Զ�˵���Ԫ����Ϊ���ų�ͨ���о���ƺͲ����������DZ�ڵ�ƫ���������ռ�����������Ŀ������ݣ�27��28��������������ê��Ⱦɫ��ѭ������DNM�ֲ��ڲ�ͬ���о���ͼS2���ơ��ƿ��ײ���P = 0.91�������ǽ����ǵ� OCD �����������������ݽ����˱Ƚϣ�ͨ�����γ������Ժ���һȾɫ�壨LOCO�������Ϻͷ��������� SE�������һ�µĸ�����û�н����о������ԣ����γ��������ͷ�����= 1.25��P < 2.2 �� 10 -16��Cochran ����P = 0.94��LOCO�����ͷ��� OR = 1.25��P< 2.2 �� 10 -16��Cochran ����P = 0.15)��
������������ DNM ���ʡ�
��A��B����A����ͬ���ܻ���������ͣ�B����ͬȾɫ��״̬��ÿ������ͻ���ʵķֲ�����Pֵ��ʾ�Ķ��ջ�ͬ���ߵ���������������ͽ���У������ʹ�üٷ����ʵĶ�����ԡ���X�൱���ɷ�����IJ��ԣ����������ORֵ�����95�������������䡣( C ) ԭ��ͼ���ۺϻ�����ѧ�鿴��ͼ��ʾ������ͨ�������ۺ�ø����Ӧ (qPCR) ��֤�� dSV��( D) �Ƚ�����ͬ���� DNM Ӱ��Ļ���IJ������ԡ�SSC ���ƣ�Simons Simplex Collection �ֵܽ��ã�pLI������� LoF ͻ�䲻���ܵĸ��ʣ�dSV����ͷ SV��
 �ڲ鿴���д�
�ڲ鿴���д�Ⱦɫ��״̬�ǿռ价���ж�����ۻ������ǵ���ϣ����Բ���������ͬ���Ļ�����Ԫ������Ϣ�����������ӡ���ǿ�ӡ�ת¼�����ƺ��ظ�������ˣ�������չ�����Ƕ� DNM �����ķ�����ͨ����ͻ��ӳ�䵽 15 �ֺ���Ⱦɫ��״̬ ( 26 )���о������ڲ���������б��ۻ������ǵķ�����������ǹ۲쵽пָ������ظ������ڷ������Ÿ��ߵ�ͻ�䣨Ⱦɫ��״̬ 8������PУ��< 0.0001 �� OR = 9.65��ͼ 1B)�����ص����鵰�ױ��H3K9me3��H4K20me3��H3K36me3Ƶ�ʸߣ��������Ƶ����Խϵ͡����������ƣ�Ⱦɫ��״̬֮��� DNM �ֲ��ڲ�ͬ�����о�֮��߶�һ�� [Pearson ���ϵ�� ( R ) > 0.96����ͼ��S2]��������Щ�о��ĸ��������һ�µģ��ӳ��������ͷ��� OR = 7.24��P= 1.23 �� 10 -10��Cochran ����P = 0.87��LOCO�����ͷ��� OR = 7.46��P < 2.2 �� 10 -16��Cochran ����P = 0.07)��
����ʶ���ͻ��Ͳ���ȱʧ�����ǻ�̽���˴�ͷ SVs (dSVs) �Ƿ���ǿ��֢�������á��ڻ���ѧϰ��⡢�ֶ�����Ⱥ��Ƶ�ʹ��ˣ����Ϻͷ�����֮������ȷ���� GnomAD Ⱥ����δ���ֵ�����DZ�� dSV�����������Ƕ����ۺ�ø��ʽ��Ӧ (qPCR)������������������֤Ϊ�����Ĵ�ͷ���塣����һ��������Ϊ�����緢��ǿ��֢�����Ի��ߣ�ID��WOC3_114_1_PT���Ļ���FBL�ĵڶ����ں��ӵ��Ӻϴ�ͷȱʧ��chr19��40331154-40331255������ S3������ dSV �����˴ֵڶ����ں��Ӻ������� 3 5' ���������������һ��������������� 2 �� 3'������FBL ������������ĸı���ͼ 1C����FBL ���� Fibrillarin������һ�ֺ˵��ף��ɼ����鵰�� H2A (H2AQ104me) �� glutamine-104 ������Ⱦɫ�ʽṹ�ͻ��� ( 29 )����һ��ȷ��� dSV ����һ�����Ի��ߣ�ID��WOC4_34����Ҳ�����緢��ǿ��֢��dSV �� 703 ������� (bp) Ƭ�� (chr12:64800102�C64800404) ���Ӻ�ȱʧ����Ӱ��XPOT�ĵ�һ���ں��ӵ�������ͼ 1C�ͱ� S3����Ԥ�ƶ����� ANNOVAR ( 30 )�Ļ��ڻ���Ĺ���ע����
������������̽������ʶ��� DNM �Ĺ���Ӱ�졣���ڻ����ͻ��������Բ�ͬ��ͨ�� LoF ������ (pLI) ( 31 )�ĸ�������������������о������ֲ��������Ƿ����ܲ�ͬͻ�����Ӱ��Ļ���֮����ڲ��죨ͼ 1D�������ǹ۲쵽���ڱ��� DNM ( P = 0.003)���ƻ��� DNM ( P = 0.03) ����� DNM ( P = 0.002)���棬OCD ��ػ������Ա� SSC ���ո�������ͻ����������ͬ�� DNM ( P = 0.25����
���⣬�����ƶ����Է����е�������Ե�ת¼�����ݽ�������Ƕ�ǿ��֢��������ѧ�����⣬��Ϊ����Ƥ��һֱ��ǿ��֢�йأ�32�������Ǽ���� Brainspan ( 33 )�ı������ݣ���ͨ����Ȩ������������� (WGCNA) ( 34 )�����˲�ǰ�Ͳ��������磬�Է�����������б����� 1 ������ı䵰���ʵ� DNM���� 1������ 2 ����������ӱ� DNM ���еĻ��� 3 ���ǵ� 1 ��͵� 2 �����ϡ�
| ��֤������֤ | λ�� | ͻ�� | ���� ���� | ͻ������ | ������ �仯 | ɸ | ���2 HDIV | CADD | ���� |
|---|---|---|---|---|---|---|---|---|---|
| WOC5_160_1 | 18:31324188 | ����ɫ�� > G | ASXL3 | ���� ɾ�� | p.F1460Lfs*5 | ������ | ������ | 35 | �ƻ��� |
| WOC3_114_1_PT | 6:29912028 | AG > AG | HLA-A | ���� ɾ�� | p.D251Tfs*45 | ������ | ������ | 32 | �ƻ��� |
| WOC5_137_1 | 1:109242401 | CAAA>CAAA | PRPF38B | ���� ɾ�� | p.K324Efs*59 | ������ | ������ | 34 | �ƻ��� |
| WOC5_26 | 2:114482963 | CA > C | SLC35F5 | ���� ɾ�� | p.L408Rfs*11 | ������ | ������ | 35 | �ƻ��� |
| WOC5_160_1 | 19:16611793 | CT > CTT | C19orf44 | ���� ���� | p.K65Efs*15 | ������ | ������ | 14.66 | �ƻ��� |
| WOC5_6 | 19:2939254 | AGTGAGGGAATGACA CCACCCTTACCCAAG GAGGCA>A | ZNF77 | ����λ�� ͻ�� ������ ȱʧ�� | p.A41Lfs*5 | ������ | ������ | 16.62 | �ƻ��� |
| WOC5_160_1 | 3:49713641 | C > T | ��̫������֯ | �ϻ� | p.Q199X | ������ | ������ | 35 | �ƻ��� |
| WOC4_28 | 13:32937534 | T > G | BRCA2 | �ϻ� | p.L2732X | ������ | ������ | 39 | �ƻ��� |
| WOC5_102_1 | 14:20779873 | G > A | CCNB1IP1 | �ϻ� | p.R224X | ������ | ������ | 37 | �ƻ��� |
| WOC4_34 | 4:47427852 | C > G | GABRB1 | �ϻ� | p.Y414X | ������ | ������ | 27 | �ƻ��� |
| WOC4_165_1 | 6:39286844 | C > T | KCNK16 | �ϻ� | p.W93X | ������ | ������ | 43 | �ƻ��� |
| WOC4_170_1 | 22:25320164 | C > T | GSM1 | �ϻ� | p.R1008X | ������ | ������ | 46 | �ƻ��� |
| WOC84 | 1:118616533 | G > A | SPAG17 | ���� | p.R777C | D | D | 29.5 | �ƻ��� |
| WOC4_170_1 | X:51640699 | C > T | ħ��1 | ���� | p.R515C | D | D | 28.9 | �ƻ��� |
| WOC5_157_1 | 5:137722015 | A > G | KDM3B | ���� | p.E362G | D | D | 28.6 | Damaging |
| WOC5_096_1 | 17:19284412 | A > G | MAPK7 | Missense | p.Y158C | D | D | 27.5 | Damaging |
| WOC5_62 | 1:36479519 | C > T | AGO3 | Missense | p.R192W | D | D | 27.4 | Damaging |
| WOC5_178_1 | 3:142741802 | C > G | U2SURP | Missense | p.P376A | D | D | 25.9 | Damaging |
| WOC4_130_1_WY | 3:9476069 | C > T | SETD5 | Missense | p.R77C | D | D | 24.6 | Damaging |
| WOC5_093_1 | 17:18195985 | G > C | TOP3A | Missense | p.P324A | D | D | 25.2 | Damaging |
| WOL3_046_1_ZXY | 19:58438675 | C > T | ZNF418 | Missense | p.G207R | D | D | 21.7 | Damaging |
| WOC5_093_1 | 1:206648328 | C > T | IKBKE | Missense | p.R32C | D | D | 28.8 | Damaging |
| WOC5_099_1 | 4:142949945 | G > T | INPP4B | Missense | p.A922D | D | D | 25.7 | Damaging |
| WOC5_149_1 | 8:131072874 | G > A | ASAP1 | Missense | p.T1048M | D | D | 24.5 | Damaging |
| WOC5_17 | 2:71817395 | C > T | DYSF | Missense | p.A1152V | D | P | 27.9 | VUS |
| WOC5_164_1 | 12:96617430 | A > T | ELK3 | Missense | p.N29I | D | P | 25 | VUS |
| WOC5_4F | 13:76381720 | G > T | LMO7 | Missense | p.R201L | D | P | 23.2 | VUS |
| WOC5_097_1 | 3:38595773 | C > T | SCN5A | Missense | p.V1586M | D | P | 23.1 | VUS |
| WOC5_130_1 | 20:33594249 | T > C | TRPC4AP | Missense | p.T606A | T | P | 25.9 | VUS |
| WOC5_161_1 | X:123519751 | G > A | TENM1 | Missense | p.T1944I | D | B | 23.6 | VUS |
| WOC5_155_1 | 11:62416110 | A > G | INTS5 | Missense | p.V481A | D | B | 23.5 | VUS |
| WOC5_078_1_X | 15:41810235 | C > T | RPAP1 | Missense | p.S1314N | T | B | 22 | VUS |
| WOC5_60 | 16:68225447 | A > G | NFATC3 | Missense | p.T959A | D | B | 21.7 | VUS |
| WOC5_091_1 | 6:32123547 | G > A | PPT2 | Missense | p.M140I | D | B | 20.8 | VUS |
| WOC5_076_1_WSH | 14:91928489 | T > C | PPP4R3A | Missense | p.M451V | T | B | 20.4 | VUS |
| WOC5_15 | 17:40149144 | T > C | DNAJC7 | Missense | p.S38G | T | B | 19.65 | VUS |
| WOC5_4F | 3:102157378 | C > T | ZPLD1 | Missense | p.P32L | �� | �� | 18.89 | VUS |
| WOC5_094_1 | 19:47207624 | G > A | PRKD2 | ���� | p.P74S | �� | �� | 16.23 | VUS |
| WOC5_138_1 | 19:36223863 | C > T | KMT2B | ���� | p.P2138L | D | �� | 15.46 | VUS |
| WOC5_116_1 | 19:57036758 | G > A | ZNF471 | ���� | p.S367N | D | �� | 15.06 | VUS |
| WOC5_152_1 | 12:113759149 | C > T | SLC8B1 | ���� | p.R54H | �� | �� | 14.72 | VUS |
| WOC5_39 | 2:217012900 | C > T | XRCC5 | ���� | p.T524I | �� | �� | 14.16 | VUS |
| WOC5_39 | 9:125551863 | A > C | OR5C1 | ���� | p.I218L | �� | �� | 12.96 | VUS |
| WOC5_084_1_PS | 6:151671185 | T > A | AKAP12 | ���� | p.D455E | �� | �� | 6.24 | VUS |
| WOC5_150_1 | 18:76753591 | G > C | SALL3 | ���� | p.V534L | �� | �� | 0.008 | VUS |
| WOC5_091_1 | X:114468473 | CCCGCCGCCGCCGCCGCC> CCCGCCGCCGCCGCC | LRCH2 | ������ ɾ�� | p.G44del | ������ | ������ | 16.51 | VUS |
�� 1�����о��м��������иı䵰���ʵ� DNM �б���
VUS�����岻ȷ���ı��塣�����ã������á�
�������ȹ۲쵽��ǰ�����е�ģ�� M16 ������ set1 �������Զ���Ԥ�ڣ�˫�� Fisher ��ȷ����P = 0.00012�������� Metascape��ͼ 2A�ͱ� S4����M16 �Ĺ��ܷḻ�������鵰�����Ρ���ԪͶ����̬������ͻ����֯����̽�� M16 �� DNM �������ͨ��ʱ�����ǹ۲쵽���� DNM ����䵱��Ŧ����ASXL3 [���� = 161��kME��ģ������ͨ�ԵĶ�����= 0.78]��SETD5������ = 45��kME = 0.75������KDM3B������ = 246��kME = 0.87������ M16 �У�ƽ������Ϊ 41�������������ǹ۲쵽�����������е�ģ�� M11 ������ set1 �������Զ���Ԥ�ڣ�P = 0.007����M11��Ҫ���������Ŵ�������ع��ܣ��繲��Ⱦɫ�����Ρ��鵰�������������Ⱦɫ�����ܣ�ͼ2B�ͱ�S4�������о� M11 �� OCD DNM �������ͨ��ʱ�����ǻ����������� OCD ����SETD5������ = 26��kME = 0.92����KDM3B������ = 27��kME = 0.84����������Ϊ M11 �е���Ŧ��ƽ������9. Ȼ��������û�������������й۲쵽 set2 �� set3 ��������ƽ����
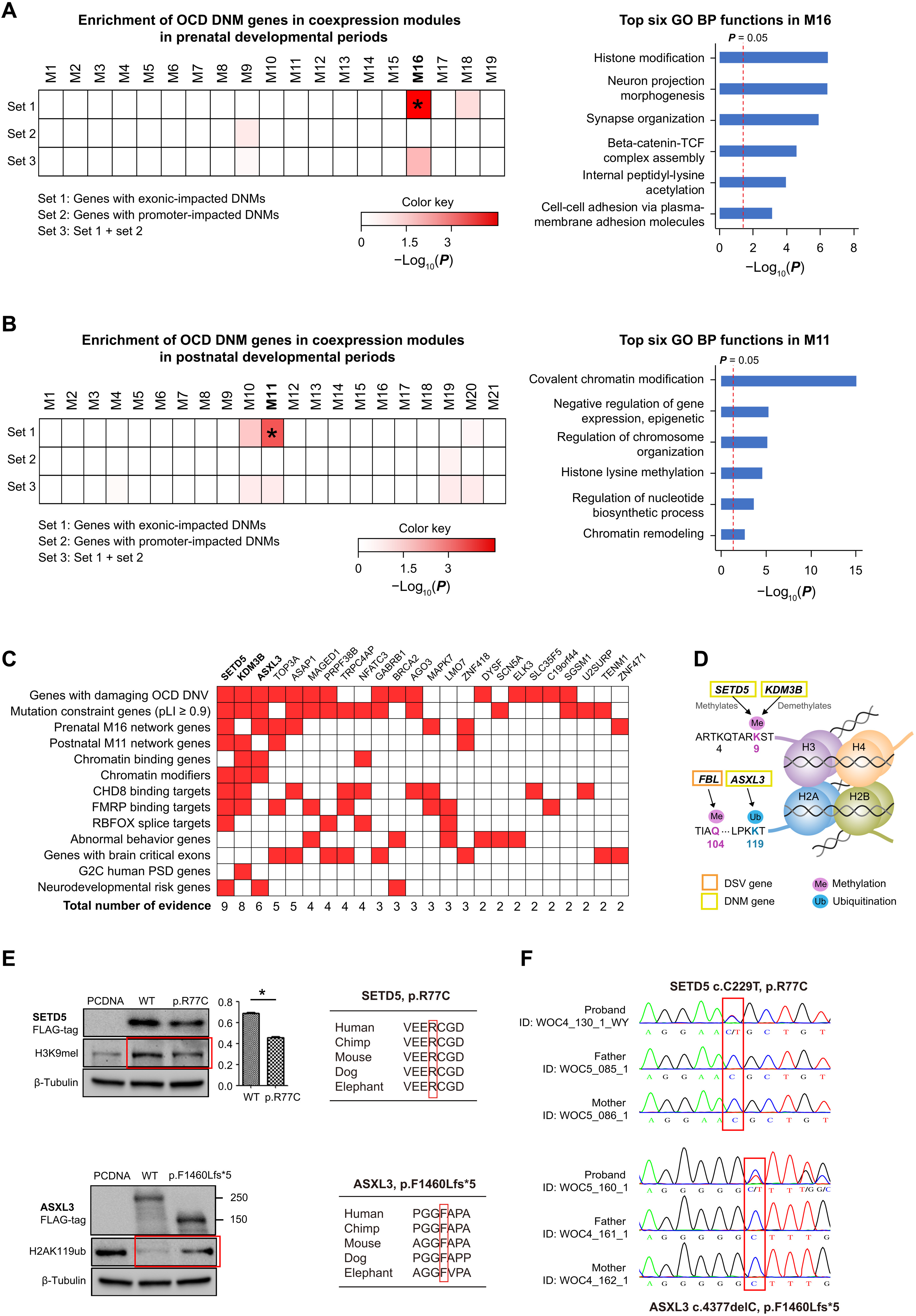
ͼ 2������ DNM ������ѧ���塣
��A��B���ڣ�A����ǰ�ͣ�B������������ԣ��ͻ������ۣ�GO��������̣�BP���Ĺ�����ģ���и�����DNMӰ��Ļ�������Ӧģ���Metascape����������ң� ������ͼ��ʾ�����̶���Pֵ��( C )����DNMӰ������DZ���²��Ե��ۺϷ�����ʹ�� 13 ��������֤����Դ���� DNM Ӱ��Ļ����������( D ) SETD5��KDM3B��ASXL3��Ⱦɫ�ʵ��ڻ���ʾ��ͼ��( E��������ӡ��������ͷ���ͻ��λ��Ŀ����ֱ������ң���( F ) (E) ͻ��� qPCR ��֤��CHD8��Ⱦɫ�ʽ���ø DNA ��ϵ��� 8��FMRP������ X �������µ��ף�RBFOX��RNA ��� fox-1 ͬԴ�� 1��G2C����֪����Ļ���
��ǰ���������Ƕ�ͻ�主����dSV ��λ�ͱ��� DNM ������ķ���ʼ���漰 OCD �е�Ⱦɫ�������Ⱦɫ���������;������ˣ����Ǽ�����ͻ����ʾ��;����ص� DNM �����Ӽ������� OCD ����ء���ˣ��������� 13 ��������֤����Դ����λ�� Jaccard ������ָ�� = 0.0244�������� 45 �� DNM ������ 1������������������ͼ 2C��������Ϻͷ�������ϸ��Ϣ�������Ƿ�������������SETD5��KDM3B��ASXL3 ������������֧��֤�� (��6) ���Ҷ���ͻ�䲻���� (pLI �� 0.9) ��Ⱦɫ�����μ������dz䵱�������ࡣSETD5���鵰��ת��ø�����ò����鵰�� H3 ��Lys 9����������KDM3B��һ���鵰��ȥ����ø������ȷ�����鵰�� H3 ��Lys 9ȥ���������⣬ASXL3�Ƕ���������ȥ����ø (PR-DUB) �������һ���֣��ɽ��鵰�� H2A ������119ȥ���ػ���ͼ 2D�������⣬�����ܻ��� WOC3_114_1_PT ��ͷ�ṹ����Ӱ��Ļ���FBL����ǰ���о��Ѿ�֤ʵ���ܹ������鵰�� H2A �Ȱ�����-104 �����ں��ʻ��ԣ�ͼ 2D����
Ϊ��ȷ����Щͻ���Ƿ���Ԥ������Ӱ�������Ⱦɫ�������еĹ��ܣ�����ѡ����SETD5��ASXL3����������̥�� (HEK) 293T ϸ���н���Ұ���� (WT) ��ͻ����ʵ������֤���裨���Ϻͷ����������������� WT/ͻ���� SETD5 �� ASXL3 �����˱����������� FLAG-tag �ںϣ����� HEK293T ϸ���Ĺ�����ʵ�顣�������������ñ���� pcDNA3.1 ���壨��Ϊ���գ���WT SETD5 �� ASXL3 �� p.R77C SETD5 �� p.F1460Lfs*5 ASXL3 ������תȾ HEK293T ϸ�������Ǵ�ÿ��ϸ�����ռ��������ѽ������ 48 Сʱ��ͨ��������ӡ������鵰�� H3K9me1 �� H2AK119ub ˮƽ�����Ƿ�����תȾ�� pcDNA3.1 ����� WT SETD5 ��ȣ�p.R77C SETD5 ͻ�����תȾ���ܱ����鵰�ױ�� H3K9 �ĵ�����ˮƽ������ p.ͼ 2E )��ͬʱ�����ǹ۲쵽�� pcDNA ������ȣ�תȾ WT ASXL3 ������ H2AK119 ���ػ�ˮƽ��֤ʵ�� WT ASXL3 ���鵰��ȥ���ػ����ܡ��෴������p.F1460Lfs*5��ASXL3ͻ�����ϸ����H2AK119���ػ�ˮƽ��pcDNA�������ƣ�������ͻ����ȱ���������鵰��ȥ���ػ����ܣ�ͼ2E����
ǿ��֢��������������;���ĸı�����ģ�����Ȱ���ϵͳ����Ͱ�ϵͳ��Ѫ����ϵͳ����ˣ����Ǽ����ڱ��о��У�����ȷ��ΪȾɫ�ʸ��Լ�SETD5��KDM3B��ASXL3��FBL����ñ����Ŵ����ε������ʱ��Ϊ���о�ǿ��֢��Ⱦɫ�����μ�������֮��ı�����أ�����ʹ���� Jaffe�������е�һ���о����Ի������������( 35)�����а�����ǿ��֢�����ͷǾ����շ����������Եı����ǰ��ҶƤ��ı����ס��������ȼ�����ǿ��֢�����Ͷ�����������Ⱦɫ�����μ�������֮����Ƥ��ѷR s��Ȼ������ͨ������ǿ��֢���ߺͶ�����֮��ľ��Թ�������죨|��Co-exp| = |R OCD �C R����|����������������ڣ���Co-exp ����ͼ 3A�ͱ� S5�������Ƿ���KDM3B�Ͷ�Ͱ�����֮������干������OCD �����Ͷ���֮�����Ÿı䣨Wilcoxon ����P = 0.025�������֮�£�����֮��Ĺ�����ASXL3�Ȱ����Լ���ASXL3��Wilcoxon ����P = 0.047����Ѫ����֮�䱻���ƻ���Wilcoxon ����P = 0.045��ͼ 3A����
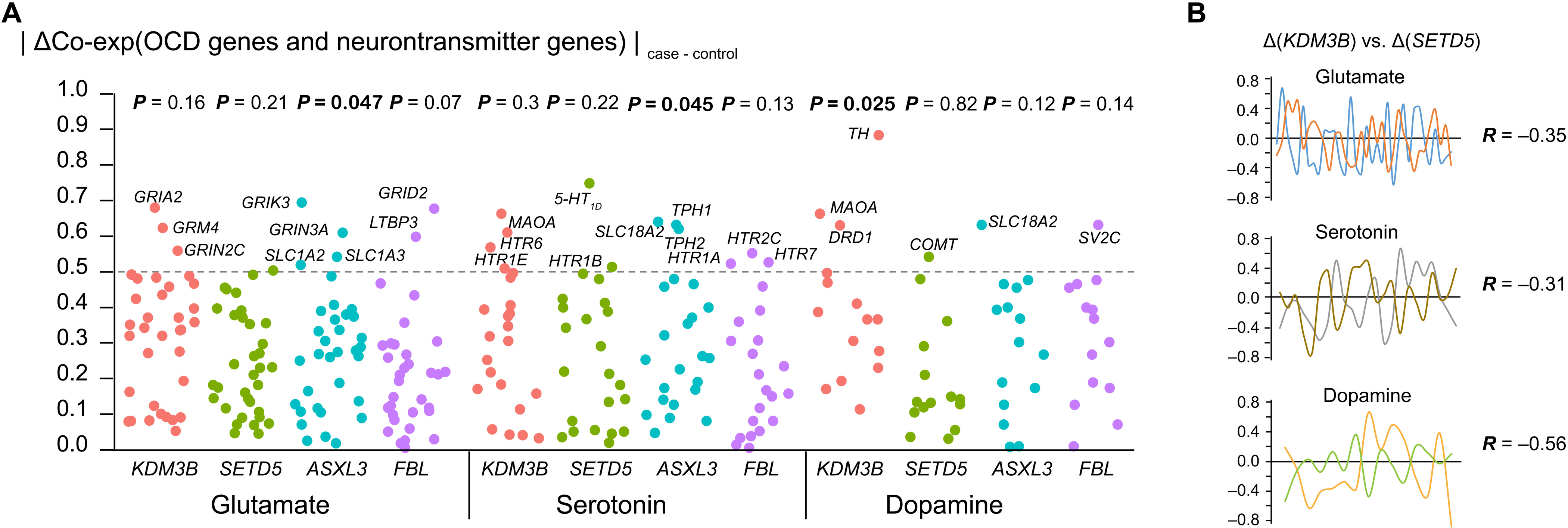
ͼ 3���ؼ���ͷ����֮��Ĺ������жϡ�
( A )ǿ��֢����ǰ��ҶƤ����FBL��SETD5��KDM3B��ASXL3�����ʣ��Ȱ��ᡢѪ���غͶ�Ͱ�����ػ���Ĺ����ÿ�������һ������һ������ϵͳ�Ļ���Y����ʾ���ߺͶ���֮�乲���� (��Co-exp) �ľ��Բ��졣ʹ�� Fisher �������Pֵ��ָʾ ��Co-exp �Ƿ����Ŵ���Ԥ�ڡ�( B ) ��KDM3B��SETD5������ж������෴�����ơ�����ÿ������ϵͳ����R��SETD5��KDM3B��ֵ���ͼ�����������ǵ�����ԡ�
ͨ�����������ǿ��֢����֮�乲����仯�������ʣ����ǹ۲쵽�����Ұ���ת��Ϊ��Ͱ����Ұ����ǻ�ø ( TH ) �����ܵ�KDM3B ���ƻ��������KDM3B = 0.89������R = 0.22 ���Ƶ�R = -0.66 ǿ��֢��P = 0.0065��ͼ 3A�ͱ� S5���������ֲ���Ѫ���غͶ�Ͱ���л��MAOA������ǿ��֢�����б�KDM3B �ƻ����� KDM3B = 0.66����R = -0.07 ���Ƶ�R= 0.59 ǿ��֢��P = 0.06����֮��Ĺ�����SETD5��Ѫ���ػ���HTR1D��Ҳ��Ϊ5-HT 1D���������ı䣨�� SETD5 = 0.75��? =����0.35?- [R = -0.4��OCD��P = 0.002����FBL���������ڷ���ϸ�����ڵ���Ͱ�����SV2C֮��Ĺ�����������ű仯���� FBL = 0.76���������е�R = -0.23 ��OCD �е�R = 0.53����
����SETD5�����鵰�ױ��H3K9����KDM3Bʹ�鵰�ױ��H3K9ȥ������ͼ 2D�������Ǽ���SETD5��KDM3B�����Բ�ͬ��ʽ������ͬ���ʵı������Ԥ�ڵ������������ڹȰ��ᡢѪ���غͶ�Ͱ�ϵͳ���۲쵽��Co-exp SETD5�� ��Co-exp KDM3֮�����R s �ֱ�ΪR = -0.35��-0.31 �� - 0.56��ͼ 3B��������෴��ASXL3����ٽ��鵰�ױ��H2A119����ͬ���鵰�ױ�ǵ�ȥ���ػ���������һ���SETD5���κ����ʵ���ϵͳ�е�KDM3B��
OCD �� TD ���ֳ���������֮���������Ŵ�ѧ�����ͺ����в�ѧ ( 36 )����ˣ�Ϊ�˽�һ���˽� OCD �ĸ����Ŵ�ѧ������ͨ������������ DNM��������Ϻͷ��������ȽϺͶԱ� OCD �� TD��ͨ��ֱ�ӱȽ� OCD �� TD ֮����ص�����ϵ�ǰ�о����ѷ����������������� ( 10 , 11 , 18 �C 20 )�����ǹ۲쵽��Щ����֮��Ļ����ص���Ԥ�ڵ�Ҫ�ࣨ���������� DNM��P = 1.21 �� 10 -38��LoF DNM��P = 9.19 �� 10 -18������ DNM��P = 1.01 �� 10 -34) (ͼ 4A )���������������о��� 15 ��������ػ����飨������Ϻͷ������� OCD �� TD ����ĸ����Ƿ�Ҳ���ơ�����ͨ���Ƚ� log 10ת���ĸ��� OR��ͼ 4B���۲쵽���ּ����ĸ���֮������ǿ����أ�Pearson R = 0.54��P = 0.01 ������������֮�乲�������������ѧ;���ϰ���

ͼ 4����OCD��TDA����ͻ��Ӱ��Ļ���Ƚϡ�
��һ��ά��ͼ��ʾ���� TD DNM Ӱ��Ļ������������о�������ǿ��֢ DNM Ӱ��Ļ���֮��Ĺ�ϵ [���� Cappi���˵�DNM ��( 10 , 11 ) �͵�ǰ���о�]��( B ) TD DNM ����ǿ��֢ DNM Ӱ��Ļ���Ļ�����֮�������ԣ��ⲻ���ɹ��ܻ���֮��������Ի�����������ģ�ƽ�� Jaccard ������ָ�� = 0.025����ÿ������� 15 ���ֶ����Ļ�����֮һ��������Ϻͷ���������X������û������ڵĻ�TD��DNM��������?���ʾ��OCD DNM-��Ӱ��Ļ���Rs ��Pֵʹ�� Pearson ��ط������㡣��C��D��TD��OCD�����ڣ�C�������ͣ�D����ϸ�������еIJ�ͬ����ģʽ�����߱�ʾ���Pֵ��ֵ (0.05)��TC���ҶƤ�ʣ�STR����״�壻PFC��ǰ��ҶƤ�㣻PC����ҶƤ�㣻OC�����Աڣ�MD�������б��ˣ��Źؽڣ�������DTH���������ԣ�CB��С��Ƥ�ʣ�AMY�����ʺˡ�
����ǿ��֢�кܴ�һ���� (75%) �� DNM ������ TD ���������Ǽ������ֲ�����ܲ������ܲ���ǿ��֢�������Ƶ��ض�����ʱ�䡢�����������Ԫϸ����ʹ�����Է����ںʹ��������BrainSpan ���� ( 33 )�����ǹ۲쵽 OCD ͻ������ڱ������� [EWCE ( 37 ) P = 0.034]���� TD ͻ���������ҶƤ�� ( P = 0.039��ͼ 4C )������ Dronc ( 38 ) �Գ�����ϸ�������������е�һ���о������� OCD DNM �������ν���ϸ�� ( P= 0.023���������� TD ͻ������� GABA ����Ԫ�и�����P = 0.006��ͼ 3D�������ݴ��Է���ʱ��û�й۲쵽�������졣��Щ�������������ȷ����ͬ�ķ�չģʽ�������ּ������ֿ�����
���о����ڶ�ʶ�������������Ʊ���ij�������DZ�ڵĸ߶��������Ŵ��������Ȥ�������� ( 14 , 15 , 17)���������״�ͨ��WGS����ȫ������ DNM ���������˻���ǿ��֢�ļ��塣����ȷ���� 4147 ��ȫ������ DNM������ 57 ��Ӱ����֤�ߵ�����������ͨ��̽��ǿ��֢��֤���� DNM ��ȫ�������ǹ۲쵽��������ê����Ⱦɫ�ʻ����鵰�ױ��Ƶ�ʸߵ���������ͻ���������Ԥ�ڣ����� H3K9me3��H4K20me3 �� H3K36me3��Ⱦɫ��״̬8�����ڼ���� OCD ͻ��ֱ��Ӱ��Ļ��������ʱ�����ǹ۲쵽 DNM ����� SSC �������Ը�������ͻ�䣬���� DNM ������ OCD ����ҪΣ�����صļ���һ�� ( 10 , 11 )��
ʹ�����Է����е�������Զ������ı������ݣ����Ƿ��������ǵ�ǿ��֢ȫ������ɸ���м����� DNM ����ֱ��ڲ�ǰ�Ͳ���������ʾ���߶���صĻ������ģʽ����ǰ�ι������������һ����һ�ĸ���ģ�� M16�����鵰�����κ���Ԫ��ͻ����֯��أ�������ΰ���һ����һ�ĸ���ģ�� M11����Ҫͻ��Ⱦɫ�����Ρ���Щ�������������εķḻģ�鶼��Ⱦɫ�ʵ���ȷ��Ϊ��صĶ���;����ǿ��������ǿ��֢�е���Ҫ�ԡ���Щ��������������� M16 �����������ӵ���Ԫ���ܣ�����ڲ�ǰ���������У�����Ĺ��ܻ������ǿ��֢�ķ�չ��
Ⱦɫ�����λ�������������ϰ��ķ������ ( 39 , 40 )���ڱ��о��У����ǹ۲쵽������ OCD �о������ѭ֤�����Ļ���SETD5��KDM3B��ASXL3����һ�����ƻ��� dSV ( FBL )�ƻ��Ļ�������Щ������Ҫ��Ⱦɫ�����μ������ܵ���ͷ�ƻ���ͻ���Ӱ�죬�������ǶԵ����ʸı������м��ߵIJ������ԡ�SETD5��KDM3B������H3K9�鵰�����������Ⱦɫ��ά��������Ҫ��41�����������ǵĹ۲���һ�£��� OCD ������Ⱦɫ������Ⱦɫ��״̬ 8���к��и����ͻ�䣬���а�����Ƶ�ʵ� H3K9 �鵰�ױ�ǣ�42�����ڵ�Ƶ�е�ͻ��SETD5�Ѿ���ASD����ǣ��39����ID��40����SETD5 �����������С��ģ�����ֳ���֪�ͼ��������Լ���Ϊ����� ( 43 )������������� OCD �ٴ��������ơ�KDM3B��һ����Ҫ���鵰�����������ø (KMT) ���鵰��������ȥ����ø (KDM) ��һ���֣����Dz��������غͱ��KDM3B��ͷ���Ŵ����²��Ա����ɵ����� ID�����İ�С���沿����Ϊ�������ۺ��� ( 44 )������ASXL3����OMIM?�ţ�615485����߲�����-Ropers�ۺ����������ϰ��������������ӳپ����˶����������ص�ID������45�����û������һ�ֶ����鵰�ף����� PR-DUB �������һ���֣�����ȥ���ػ� H2AUb1 �Ĺ��ܣ�45������ˣ���ǰ���Ŵ�ѧ�о��Ѿ�ͨ����ͬ�ı����Ŵ����ػ���ȷ����������������������ϰ�����Ҫ��ϵ��FBLͨ���鵼�鵰�� H2A (H2AQ104me)�ġ�Gln 105 ���������䵱�����ʼ�ת��ø���������λ��� FACT ������Ľ�� ( 29 )�����������е� mRNA �͵������б��� ( 46 )��pLI ͻ�䲻�������� > 0.95������FBL�뾫��֮��û��ֱ����ϵ����֮ǰ���о��ѽ�FBL ��SNP�������ϵ������������Ŷ���л����ʣ�47)����������������о����г����Ϊ�ļ�����DZ�ں�ѡ�ߡ�
Cappi���������������о���( 11 ) ��Ⱦɫ�����ܻ���CHD8������Ⱦɫ������ø DNA ��ϵ��� 8������Ϊ OCD �Ͷ������������ķ��ջ��� ( 48 , 49 )���������ڸ��Ӽ����Ķ�������ʺ����ǵ����������������о���û����CHD8������ DNM�����ѷ���SETD5��KDM3B����CHD8 ����ϰ���( 49 )����ˣ���֤ʵ��Ⱦɫ�ʵ��ڲ���ǿ��֢�������Ƶĸ��
ǿ��֢�IJ�������ѧ��Ƥ��-��״��-����-Ƥ�� (CSTC) ��·���쳣�Լ��������йȰ��ᡢѪ���غͶ�Ͱ�ϵͳ��ʧ���йء������Pauls���������һ�������˵�·����ѧ���Ŵ�/�����Ŵ�Ԫ�ص� OCD ģ����( 4)������������Ŵ����յ�ǿ��֢���߿��������ܵ��������ص�Ӱ�죬��Щ�������ؿ���ͨ�������Ŵ����ƴ����Ȱ��ᡢѪ���غͶ�Ͱ�ϵͳ��ػ���ı������Ρ����ǵķ�����������ǿ��֢���ߵ�ǰ��ҶƤ���У�����ϵͳ�����Ⱦɫ�����μ�֮��Ĺ�����ģʽ���������Ÿı䡣��Щ����������漰SETD5��KDM3B��ASXL3��FBL ��Ⱦɫ�����������ǿ��Ʊ�Ҫ������֪���ܵ�����ϵͳ��������ε��ڼ������ּ������κβ��ֵ��ж϶����ܵ����쳣��ǿ�ȱ��͡�
�����ڵ�ǰ�о���ֻ�� 53 �����Ӽ�ͥ�������ǵ��о��������һ�µĹ۲�֮һ��Ⱦɫ�����ι��ܵķḻ������� OCD �ı����Ŵ����ؾ��и߶����š�������֤�ݣ������� OCD ����DZ�ڵı��ۻ���������ԡ���ˣ�����ͻ������ͨ������Ϊ��ǿ��֢�ķ��������йأ�8�������ǵ��о����ǿ���˳���ͻ�������������ƵĴ��ڡ�������Ҫ��һ���Ĵ��ģ�о���֤�� DNM �� OCD �еı����Ŵ����ص��²����ã������ڱ��о��е��е���������ͳ��ѧ���壬���ǵĹ۲����ǿ��ŵģ����ǵ� ASD Ҳ�����Ƶķ��֡��� SCZ���Լ�CHD8�������ǿ��֢���������о��б�ȷ��Ϊ����Ҫ�ĺ�ѡ���գ�����Ҳ��Ⱦɫ�����ܻ���
������ǵ��о�����ṩ��֤�ݣ�����ǿ��֢�� TD �к�ǿ���Ŵ��������������������У�ת¼ģʽ������������Ԫϸ�����Ͷ��졣���� TD �� OCD ��������������Ͳ�ǰ��Ҳ�۲쵽���Ƶĸ������������ʴ�л ( 50 ) ��֢״�������쳣һ�������ǵ��о������һ��֧�������ּ������Ƿ����ϰ���11����Ȼ����ǿ��֢�����ڱ��������б��ֳ����صĸ߱������ǿ��֢����ģ���е� CSTC ��·һ�¡�TS��OCD֮���������ͬ�ͷ�����ԴӶ������յĽǶ������͡����������ּ������ڹ㷺���Ŵ��ͱ��������ԣ������ڱ��о��н������ DNM�����ǵ��о�������ܽ����������ּ�����������/��ɢ�����һС���֡�������ˣ����ǵķ�������Ϊ���� OCD �� TD ֮��DZ�ڵĺϲ�֢�ṩ����Ҫ��һ����
����ȫѪ������ DNA �Ŀ����Ժͱ�����Ϣ�������ԣ������ռ��� 53 ������ص����Ӽ�ͥ������ 160 �����塣���Ӽ�ͥ�� 52 �������飨ÿ����ͥ��һ����ǿ��֢Ӱ��ĺ�����ܹ� 156 ����������һ����������ǿ��֢Ӱ��ĺ���ļ�ͥ���ܹ��ĸ���������ɡ��� S1 ���ṩ��ÿ�������ߵ���ϸ��Ϣ��������ǿ��֢Ӱ��Ļ��߾�����������������i�����ݾ�����Ϻ�ͳ���ֲ���İ棨DSM-IV����51�������߱����Ϊ����ǿ��֢���� ����(ii) ������ 18 �� 65 ��֮�䣬���� (iii) Ү³-����ǿ��֢�����ֽܷ�ֵֹΪ 16��������� (i) ������ DSM-IV ����������������������ų�����ǿ��֢��(ii) ���ж����ضȵ���ɱ���(iii) �ǻ��л����ڵ�Ů�ԡ����о��ų��˱���ϻ��� SCZ����������ϰ���ASD��δ����ָ�����ձ��Է����ϰ��� ID �Ļ��ߡ�ͨ��ʹ�� Mini International Neuropsychiatric Interview ( 52)�������ų��˾����κ� DSM-IV Axis I ����ѧ��ϵĸ�ĸ�ļ�ͥ�����Ϻ��о����������Ļ������ίԱ���������в����߾����֪��ͬ�⡣
��ȫѪ���ܰ�ĸϸ����Դ��ϸ��ϵ����ȡ�Ļ����� DNA ͨ�� PicoGreen ��������Ӿ����������Ȼ���� Novogene��Novogene Biosciences Inc.���������й�������DNA ����ͨ�� Qubit 3.0 ���������� 1 ��g δ����Ļ����� DNA ���ڻ������Ŀ��Ʊ��� WGS�������� Illumina HiSeq 4000 ����ƽ̨��150 bp ˫ĩ�˶������϶�֮ǰ��δ������������������������˲���ʹ�ô������̣����д�ͷ���嶼��ȷ��Ϊ��������Ȼ��ʹ�� Integrative Genomics Viewer (IGV) ͨ����������Ŀ��ӻ��ֶ���� ( 53 )��ʹ�� wANNOVAR ( 30�������ǵ� WGS ���ݵ�ƽ���������Ϊ 29.9 ����ƽ��������Ϊ 99.87%����һ�������౻�ų�����������֮���ʣ�µ� 53 �������౻�ύ���к���������
ʹ�� Sentieon DNAseq ( 54 ) �����ܵ���ʹ�� BWA-mem ( 55 ) v0.7.15-r1140�������� GRCh37.63 ���������ο��������룬��ͨ�� Sentieon Dedup ��������Ȼ���ظ���ȡ�� Sentieon LocusCollector ��Dz�ɾ����ʹ�� Sentieon Realigner �� QualCal ִ�� indel ����У�ͻ���������������У��������������ʹ�� SAMtools���汾 1.3.1��flagstat��56���� Picard �� WGA ָ�꣨57�����汾 2.5.0���� BAM �ļ����������������
ʹ�������㷨����ϵ��ô�ͷ SNV �Ͳ���ȱʧ��SAMtools bcftools���汾 1.3.1����56���ͱ�Ҷ˹��� TrioDeNovo��58����ÿ����ͥ��Ĭ�����á�����������ֵ���˼ٶ��Ĵ�ͷ���壺��i��������������30��(ii) ��֤�ߺ�ĸ˫������С�������Ϊ 15 ����ȡ�㣻(iii) ���������30��(iv) ��λ����ƽ�� (AB) < 0.05 �ĸ���ĸ�״����ӣ�(v) AB ���� 0.3 �� 0.7 ֮�����֤�����Ӻ��ӣ�(vi) TrioDeNovo �Ĵ�ͷ�������֡�7��(vii) ���עΪ�ڶ��ظ�����֪�����ص���(viii) 1000 ������ƻ��е���Ҫ��λ����Ƶ�� �� 5.0 �� 10 -3 ( 59)��ExAC ( 31 )��EVS ( 60 ) �� gnomAD ( 61 ) ���ݿ⣻(ix) �� 50 bp �����ھۼ��ı����ѱ�ɾ��������� silico BLAT ���� ( 62 ) ��ʹ�����Լ���ıȶԶ���������д�ͷ������Ȼ��ʹ�� IGV ͨ���ȶԶ����Ŀ��ӻ��ֶ���� ( 53 )��
�������ĸ���ͬ��ע����ע�����£���1���������ͣ�ÿ������ʹ�� wANNOVAR��30����ͻ�����ͷ��������� SNV �� indel��<50 bp�����������롢ֹͣ���棨�ϻ�������ͬ��SNV�����壩��ͬ��SNV�ȣ�2��������λ��ע�ͣ�����Ļ�����λ����wannovar��30��ע���������������ȼ������롢�ں��ӡ�UTR�����Ρ����Ρ�ncRNA�ͻ���䡣(3) ��������Է�����������صĻ����б�ѡ�����£� (3.1) CHD8�л�����Ϊ��������Ⱦɫ�����߳��������о����б������� ( 49 , 63����(3.2) FMRP�л�����Ϊ���� Darnell�������б���������( 64 ) �Ͱ�˹��ŵ���ˡ���65����(3.3) RBFOX����Ŀ��ѡ��Weyn -Vanhentenryck ���ˡ���66����(3.4) �� Genes2Cognition ���ݿ� ( 67 )����ȡ����ͻ�����ܶȵ�����(3.5) �������� ExAC ���ݿ��б�����Ϊ pLI ���� �� 0.9 ( 31 )��(3.6) ����Ⱦɫ��������Ļ���� Chen����������( 68����(3.7) �����ϰ����ջ������� Stessman���ˡ���69����
�Ѿ�ʹ���������͵ķ�������� SV��Manta ( 70 )��cn.MOPS ( 71 )��DELLY ( 72 ) �� LUMPY ( 73 )������Ӧ���� SV2 ( 74) �� 50 bp �� 10 Mb �ڵ� SV �ϲ���ÿ������� VCF �ļ��У���ѡ�� FILTER ��Ǻ� DENOVO_FILTER ����ж����С�PASS���� SV ��Ϊ��ѡ dSV�����⣬���Ƕ����� dSV ִ�в�ͬ�Ĺ����������£��������Ի��ߣ�����ɸѡ��Ⱦɫ��� X Ⱦɫ����δ��Ӱ��ĸ�ĸ�в����ڻ� Y Ⱦɫ����δ��Ӱ��ĸ����в����ڵĴ��� SV������Ů�Ի��ߣ�����ɸѡ�Ӻ�/���� SV���ڳ�Ⱦɫ��� X Ⱦɫ����δ��Ӱ��ĸ�ĸ�в�������Щ SV���� SV2 ����DZ�� dSV ͨ�����������һ�����ˡ����ȣ���������������ר�Ҽ������Ӧ��������ÿ�� dSV �� IGV����ɾ����û����Ӧ�����仯����Щ����Σ���������� GnomAD ���ݿⲢɾ���������������ֹ��� dSV�������ڷǼ���������Ⱥ�У����� 50% �� dSV ���ȱ����� GnomAD ��ͬ���� SV ���ǡ��������ͨ�� qPCR ��֤�˺�ѡ dSV �Ĵ��ڡ�
Ϊ�˼���ȫ����������������ͻ���ʣ��������ȴ� UCSC ����������������� hg19 �����������Ӻ� UTR ����ķ�Χ�����������������������г��� 20 �������ʵļ�������������顣Ȼ������ʹ��R ���ġ� t.test ���������������� 53 �� OCD ������ 95% ���������ڵ�ƽ��ͻ������Ϊ��ȷ�� OCD ���ߺͽ�������֮�� DNM �ֲ��IJ��죬�������������� SSC �ֵܽ��õ� DNM��15) �����ؼ��������о�֮����� DNM �������в�ͬ������Ӧ�� Fisher ��ȷ���������� OR���� OR ����������������ǿ��֢������ȣ�DNM ����Ա��ʡ�Ȼ�����Ǹ��� Annovar ( 30 ) ע���������б���ͻ�������ǽ��������ȱʧ����ʼλ��/ֹͣλ��/����λ��ͻ�䶨��Ϊ LoF ͻ�䣬����ͬ�� SNV ����Ϊ����ͻ�䡣
���DZȽ������� An���˵�OCD ���ߺ� SSC �ֵܽ���֮��� CADD-phred ������( 15 ) �����Ӻͻ�����ͻ�䡣Ϊ�������������ܣ������ռ�����ǿ��֢���߲�ͬ���ͱ��� DNM��LoF�����塢�ƻ��Ժ�ͬ��ʣ�Ӱ��Ļ����pLI ( 31 ) ���֡���ͨ��˫�� Wilcoxon �Ⱥͼ������ֵ������ CADD-phred ��Ӧ��ͻ�����س̶ȵİٷֵȼ����Ҳ���ѭ��̬�ֲ�������Ӧ���ۻ��ֲ����������ӻ� CADD-phred ���ֵķֲ��� Wilcoxon �Ⱥͼ������Ƚ�Ԥ������س̶ȡ�Ϊ������ GC ƫ��Բ�ͬ�����������Ӱ�죬���DZȽ��� OCD ���ݺ� SSC ������ DNM ֮��� GC �����ͻ��ȡ�GC�����ͻ��ȵ���������GenBank��Pֵͨ��˫��Wilcoxon�Ⱥͼ�����㡣
���ǽ�ǿ��֢���ߵĽ����� PsyMuKB ���ݿ��л�õ����������������ٻ���ID��SCZ �� ASD���Ľ�������˱Ƚ� ( 46 )������������һЩ�о�������õ����������ϸ��Ϣ�����Ƕ�ͨ��ͬ��ͻ�������ͻ�����Ӧ���˼� Fisher ���顣
Ϊ���о�ͻ��Ĺ��ܻ�����ֲ����������ȴ� ENSEMBL ( 25 ) �� PsychENCODE ( 24 ) ���������������鹦��������ĸ����塣): (i) ȫ���������Ǵ� biomaRt �л�������б������ķ�Χ����ĿΪ��start_position���͡�end_position����(ii) �����ӣ����� (i) �л�õ����л��������ҵ����������ת¼������Ӧ��ת¼��ʼλ�� (TSS)�����ǽ�������������Ϊ TSS ���� 2-kb ������ 1-kb��(iii) �����ӽ����������Ǵ� PsychENCODE ��Դҳ������������ê��ѭ���ļ������ļ��Ǵ�����ǰ��ҶƤ��� HI-C �����л�õġ�(iv) ��ǿ�ӣ��� PsychENCODE ��Դҳ�棬����������ʹ��ǰ��ҶƤ�� HI-C �������ɵ���ǿ��-�����ļ�������ȡ��������ǿ���������ǻ��� Roadmap ��Ŀ��26), �� chromHMM ( 75 )����ó���
Ϊ����֤ͻ��ֲ��������������ЧӦ�����ģ����Ƕ����Խ���������ӻ���Ⱦɫ��״̬ 8��Ӧ�������в��ԡ����ȣ��������ѡ���� 53 �����ղ��ظ��� Fisher ���顣�˹����ظ� 10,000 �Σ�����ʹ�� OR �ķֲ�������OR = 1��Pֵ���������ɾ����һ��Ⱦɫ���ϵ�����ͻ�䣬����ʣ���ͻ���ظ� Fisher ���顣�� 22 ����Ⱦɫ���е�ÿһ���ظ��ù��̡�
BrainSpan RNA �������� ( 33 ) Ӧ�������¹������������������ͨ����ǰ�Ͳ���������������Ϊ������ͬ��ʱ�ڼ����������д�����������ÿ���Ӽ�������������һ��������ÿǧ���ÿ����ӳ����� (RPKM) >0 �ҷ���ϵ�� >0.3 �Ļ������� WGCNA ( 34 ) ������ʣ������ݾ��� log 10 ת������ǰ�Ӽ�������ֵ����Ϊ 12�������Ӽ�����Ϊ 16����Сģ���С����Ϊ 20������ͨ������ Pearson R�� WGCNA R ���е� blockwiseModules ��������ǩ��������������Ϊ���и�߶�Ϊ 0.3 ��ģ�顣�ڼ������Ӽ��еĹ�����ģ�������ͨ�� Fisher ��ȷ���������Я�� OCD ����ͻ��Ļ����Ƿ���ijЩģ���и�����Ȼ������ģ�����ڻ����帻�������������
Ϊ��̽���ĸ��ؼ�����SETD5��KDM3B��FBL ��ASXL3������������ϵͳ���Ȱ��ᡢѪ���غͶ�Ͱ���֮���DZ��ʧ�����������ȼ����˹ؼ������ǰ��ҶƤ����������������֮���Pearson's R Jaffe���˵�����������( 35��������ʹ��˫�� Wilcoxon �Ⱥͼ��������Ⱦɫ�����μ�������֮������干������ǿ��֢���ߺͶ�����֮���Ƿ����š�Ϊ�˲鿴�Ƿ����κε������ʻ�������ʧ�������DZȽ���|��Co-exp| ÿ�������|��Co-exp|������ֲ� ��Ӧ�Ĺؼ���������һ�������ϵ�Pֵ��
SETD5-FLAG �� ASXL3-FLAG ��������������������Ƽ�����˾���й����ϣ���ʹ�� Q5 �����ձ��Լ��У�New England BioLabs������ SETD5 R77C �� ASXL3 F1460Lfs*5 ͻ��������������������ͨ�� Sanger ����ȷ�ϡ�
HEK293T ϸ���ڱ����� 1�� �ѽ��Һ�ѽ� 30 ���ӣ�Ȼ���� 12,000 rpm ��ת������ 15 ���ӡ�ͨ���� 4% �� 20% SDS-�۱�ϩ����������Ӿ�����ϵ�Ӿ����ϸ���ѽ��Ȼ��ת�Ƶ�������ά��Ĥ�ϡ��ú� 5% ţ�̵� TBST��Tris ������ˮ��0.1% Tween? 20�����Ĥ 1 Сʱ������ 4��C ������Ӧ��һ��������ҹ���ÿ�-FLAG �Ϳ�-��-�ܵ��ף�Sigma -Aldrich)���ÿ� H3K9me1 (Abcam) ���ÿ� H2AK119ub (Cell Signaling Technology)���������������� 20�� �� 25��C �£���������������ø��ǵ�ɽ���������� G (Beyotime) һ����� 1 Сʱ��Ȼ��ʹ�� ImageJ ����ͨ�����ܶȷ�����������ӡ���Ķ�����
���Ǵ� psyMuKB ���ݿ� ( 46 )������ OCD �� TD �Ĺ������õ� DNM �����������ݼ�����������ǿ��֢�о� ( 10 , 11 ) ������ TD �о� ( 18 �C 20 )����¼�˾�������һ������� LoF ͻ������л������ǽ����ǵķ�ͬ��ͻ��������ѷ����� OCD DNM �������ϣ�����������������ǰ�� TD DNM ������бȽϡ�
Ϊ�˷��� TD �� OCD ������ص�������ʹ�ñ��������б���Ϊ���е����ʱ����������˳����β��ԡ�����˫β�����Pֵ��Ϊ�˷��� TD �� OCD ����Ĺ����������������ȴ� psyMuKB ( 46 ) ������� 15 ����������б�����Ҫ�ǹ���������ϵͳ��������ͨ�� Fisher ��ȷ���������������Щ�����еĸ��� TD/OCD ����Ȼ�����Ƕ��������ּ����Ļ����-log 10 (OR)������ Pearson ��ط�����Ϊ�˷��� TD �� OCD �����ʱ�պ�ϸ�����������Ա���ģʽ������Ӧ���� EWCE R ����37����һ�����ڻ���ϸ�����������Ծ���������������ߣ����������ݼ����и�����������i��Brainspan��33���������ں������������ii��Dronc ���ݣ�38�����ڳ�����ϸ�����ͷ���������������Ӧ�������ж��壨37������������ͨ����generate.celltype.data�����������������������ݼ��������Ծ�����������ͨ����bootstrap.enrichment.test�������������� TD/OCD ����ĸ��������ǽ����������б�����Ϊ�ڲ��Ա������ݼ���ע�͵����л���




