
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
Nature发布“整个蛋白质世界”:AlphaFold预测几乎所有已知蛋白质的形状
【字体: 大 中 小 】 时间:2022年08月01日 来源:nature
编辑推荐:
DeepMind的AlphaFold工具已经确定了地球上几乎所有已知生物体中约2亿种蛋白质的结构。
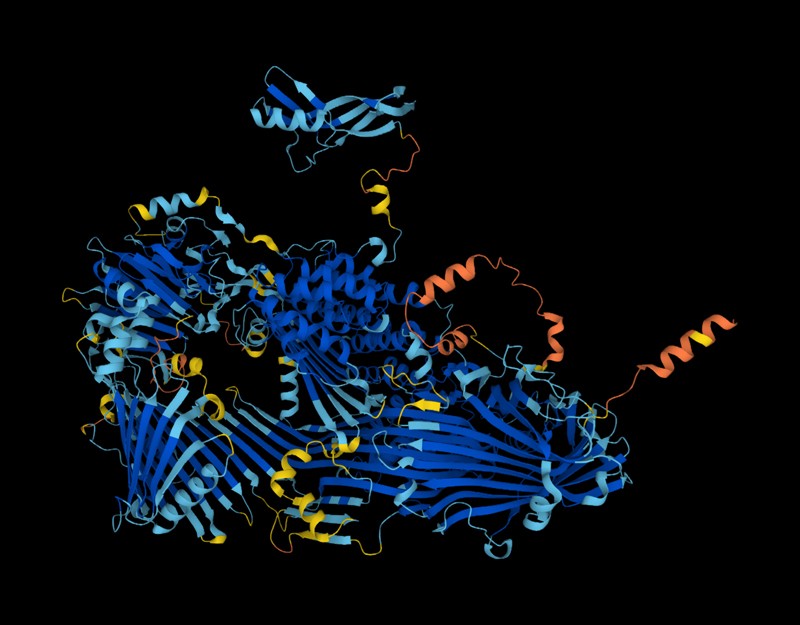
由AlphaFold工具预测的卵黄蛋白原(卵黄蛋白的前体)的结构
从今天开始,确定科学上已知的几乎所有蛋白质的3D形状将变得和在谷歌搜索中输入一样简单。
研究人员使用革命性的人工智能(AI)网络:AlphaFold来预测来自100万种物种的约2亿种蛋白质的结构,几乎覆盖了地球上所有已知的蛋白质。
这些数据将在一个由DeepMind建立的数据库中免费提供。DeepMind是谷歌旗下位于伦敦的人工智能公司,开发了AlphaFold,而欧洲分子生物学实验室的欧洲生物信息学研究所(emble - ebi)是英国剑桥附近的一个政府间组织。
AlphaFold和人工智能蛋白质折叠革命的下一个目标是什么?
DeepMind首席执行官德米斯・哈萨比斯在新闻发布会上说:“基本上你可以认为它涵盖了整个蛋白质宇宙。我们正处于数字生物学新时代的开端。”
蛋白质的3D形状或结构决定了它在细胞中的功能。大多数药物都是利用结构信息设计的,而精确的图谱往往是发现蛋白质工作原理的第一步。
DeepMind使用一种名为深度学习的人工智能技术开发了AlphaFold网络,一年前推出的AlphaFold数据库包含35万个结构预测,几乎涵盖了人类、小鼠和其他19种广泛研究的生物产生的所有蛋白质。自那以后,该目录已经膨胀到大约100万个条目。
伦敦大学学院(University College London)的计算生物学家克里斯汀・奥伦戈(Christine Orengo)说:“我们正在为这个巨大的宝藏的释放做准备。她利用AlphaFold数据库识别了新的蛋白质家族。所有的数据都为我们做出了预测,这简直太棒了。”
去年AlphaFold的发布在生命科学界引起了轰动,他们一直在争相利用这个工具。该网络对蛋白质的三维形状或结构进行高度准确的预测。它还提供了有关预测准确性的信息,以便研究人员知道该依赖哪一个。传统上,科学家们使用x射线晶体学和冷冻电子显微镜等耗时且昂贵的实验方法来解决蛋白质结构。
根据EMBL-EBI,超过2.14亿的预测中约有35%被认为是高度准确的,这意味着它们和实验确定的结构一样好。另外45%的人被认为在许多申请中有足够的信心依靠。
“它将改变一切”:DeepMind的人工智能在解决蛋白质结构方面取得了巨大的飞跃
许多AlphaFold结构已经足够好,可以在某些应用程序中取代实验结构。在其他情况下,研究人员使用AlphaFold预测来验证和理解实验数据。糟糕的预测通常是显而易见的,其中一些是由于蛋白质本身的内在紊乱导致的,这意味着它没有明确的形状,至少没有其他分子存在。
今天公布的2亿个预测是基于另一个名为UNIPROT的数据库中的序列。巴塞罗那何塞卡雷拉斯白血病研究所(IJC)的计算生物学家Eduard Porta Pardo说,科学家可能已经对其中一些蛋白质的形状有了想法,因为它们覆盖在实验结构的数据库中,或者类似于这样的存储库中的其他蛋白质。
但Porta说,这样的条目往往偏向于人类、小鼠和其他哺乳动物的蛋白质,所以AlphaFold可能会增加重要的知识,因为它从许多更多样化的生物中提取。“这将是一种很棒的资源。一旦它发布,我可能会马上下载,”Porta说。
因为AlphaFold软件已经问世一年了,研究人员已经有能力预测他们想要的任何蛋白质的结构。但许多人说,在单一数据库中进行预测将为研究人员节省时间、金钱和精力。“这是你要消除的另一个进入障碍,我用过很多AlpahFold模型。我自己从来没有运行过AlphaFold。”
德国汉堡EMBL的结构建模师扬・科辛斯基(Jan Kosinski)在过去一年中一直在运营AlphaFold网络,他等不及数据库的扩张。他的团队花了3周时间来预测病原体的蛋白质组――生物体所有蛋白质的集合。“现在我们可以下载所有的模型,”他在发布会上说。
在数据库中拥有几乎所有已知的蛋白质也将使新的研究成为可能。奥伦戈的团队已经使用AlphaFold数据库来识别新的蛋白质家族,他们现在将在更大的规模上进行这项工作。她的实验室还将利用扩大的数据库来了解具有有益特性的蛋白质的进化,比如消耗塑料的能力,或者令人担忧的蛋白质的进化,比如可能导致癌症的蛋白质。在数据库中确定这些蛋白质的远亲可以确定它们特性的基础。
首尔国立大学(Seoul National University)的计算生物学家Martin Steinegger帮助开发了基于云的AlphaFold版本,他对数据库的扩展感到兴奋。但他说,研究人员可能仍然需要自己运行这个网络。人们越来越多地使用AlphaFold来确定蛋白质之间的相互作用,而这样的预测并没有出现在数据库中。通过对来自土壤、海水和其他“宏基因组”来源的遗传物质进行测序也无法确定微生物蛋白。
Steinegger说,扩展后的AlphaFold数据库的一些复杂应用程序也可能依赖于下载其全部23tb的内容,这对于许多团队来说是不可行的。基于云的存储也可能成本高昂00作开发了一个名为FoldSeek的软件工具,它可以快速找到结构相似的蛋白质,应该能够大幅压缩AlphaFold的数据。
即使包含了所有已知的蛋白质,随着新生物的发现,AlphaFold数据库也需要更新。随着新的结构信息的出现,AlphaFold的预测能力也会提高。哈萨比斯表示,DeepMind已经承诺长期支持数据库,他可以看到每年的更新。
他希望AlphaFold数据库的可用性将对生命科学产生持久的影响。“这需要在思维上做出相当大的改变。”

