
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
我国学者在非绝热全量子化动力学理论方法研究中取得进展
【字体: 大 中 小 】 时间:2023年06月09日 来源:国家自然科学基金委员会
编辑推荐:
研究成果以“含有电子跃迁的路径积分动力学(Ring Polymer Molecular Dynamics with Electronic Transitions)”为题,发表在《物理评论快报》(Physical Review Letters)上
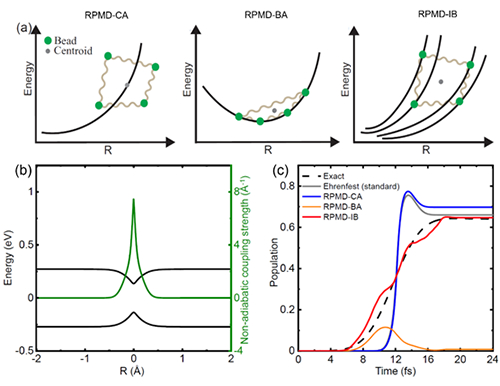 |
图 几种非绝热量子动力学方法及其与量子力学严格解的比较:(a)非绝热路径积分分子动力学方法图示;(b)二能级系统的模型势能面(黑线)与非绝热耦合量(绿线);(c)通过势垒发生电子跃迁时,不同方法计算得到的电子在高能级的占据数随时间的变化。虚线为量子力学严格解。红线为本工作提出的RPMD-IB新方法
在国家自然科学基金项目(批准号:12025407、11934003)等资助下,中国科学院物理研究所/北京凝聚态物理国家研究中心孟胜研究员课题组在非绝热全量子动力学理论方法研究中取得进展,提出一种基于路径积分分子动力学的非绝热动力学方法,可同时描述凝聚态物质中原子核量子效应和电子跃迁效应。研究成果以“含有电子跃迁的路径积分动力学(Ring Polymer Molecular Dynamics with Electronic Transitions)”为题,发表在《物理评论快报》(Physical Review Letters)上。论文链接:https://doi.org/10.1103/PhysRevLett.130.166401。
目前,基于玻恩―奥本海默近似的绝热动力学方法和考虑经典点粒子近似的混合量子―经典动力学方法是最主流的两种分子动力学模拟方法。然而,在处理非绝热现象和原子核量子效应并存的物理问题时,这两种方法都存在一些局限。例如,在解决具体物理问题时,由于真实材料中原子数目众多,导致计算量庞大,这些现有动力学方法都难以与常用的第一性原理计算方法相结合来描述真实材料的动力学特征。因此,发展一种计算量可控且能够准确模拟真实材料中全量子效应的计算方法对解决凝聚态物理中的实际问题极为关键。
在本工作中,孟胜课题组通过严格的理论分析,并与量子力学解析解作比较,发展了一种基于路径积分分子动力学的非绝热动力学方法,能够同时描述凝聚态物质的原子核量子效应和电子跃迁效应。该方法将路径积分分子动力学与Ehrenfest定理相结合,不仅能够准确描述在体系演化过程中的原子核量子效应,同时也能够描述体系演化时电子在不同能级间的激发情况(即非绝热效应)。该方法可以与课题组自主开发的第一性原理激发态动力学模拟软件TDAP相结合,对具有数百原子的真实材料体系中的量子动力学过程进行准确模拟。为进一步证实其有效性,孟胜课题组将该方法应用于水团簇和液态水中水分子受光激发分解、二氧化钛与水界面处的电荷转移、氢离子碰撞穿过石墨烯薄膜等过程的第一性原理计算模拟中。模拟结果显示,在光激发之后的50飞秒内,水分子二聚体H2O-H2O+中完成了质子转移,计算得到的质子转移速率与实验测量结果完全吻合,体现了该方法的优越性。
非绝热动力学方法计算得到的电子跃迁几率和原子核的量子分布更接近严格解和实验观测结果,充分表明了该方法的准确性和实用性,对深入理解电子激发过程中的原子核量子效应和非绝热效应具有重要作用,为研究凝聚态物质中的全量子效应提供了重要的理论计算方法。

