|
2019年4月,美国太平洋生物科学公司(Pacific Biosciences,以下简称PacBio)重磅发布新一代测序系统PacBio SequelⅡ,与PacBio Sequel测序系统相比,PacBio SequelⅡ芯片支持8M SMRT Cell ,单张芯片测序通量提升了8倍,并可产出高精准度的长读长HiFi reads,碱基准确度可达99.9% 以上,具有测序通量更高、耗时更短、准确度更高、应用更灵活等特点。
|

|

|
|
1 million ZMWs
|
8 million ZMWs
|
|
SMRT Cell 1M
|
SMRT Cell 8M
|
与二代Illumina测序平台相比,三代测序平台PacBio SequelⅡ应用SMRT 测序技术实现单分子实时测序。SMRT 测序原理是以SMRT Cell为载体,每个SMRT Cell上布满了数百万个零模波导孔(ZMW),测序时DNA聚合酶和一条模板分子被瞄定在ZMW孔底部进行反应,位于小孔底部的激发光能够激发核苷酸底物上的荧光标记,进而通过监测系统将荧光信号记录下来,从而获得碱基信息。整个测序过程 DNA 分子不需要经过PCR扩增,实现了对每一条DNA分子的单独测序。
A.长读长、高产出
PacBio Sequel II 测序Polymerase reads 平均读长可达 25kb 以上,N50 在 35kb 以上,另外目前单个 SMRT Cell 产量有大幅提升,安诺基因目前CLR模式单个 SMRT Cell 的数据产量平均在 110Gb 左右,而CCS模式平均单个SMRT Cell 的数据产量位250Gb左右。
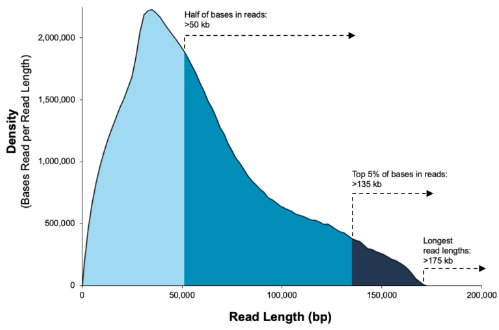
SequelII平台酶读长展示
B.高一致性准确度
PacBio SMRT 测序的原始数据错误率在 10%~12% 左右,但这种错误率是随机发生的,不存在系统偏好性,因此,PacBio测序可以利用自身的数据进行纠错,当数据深度达到 50X 左右时,一致性序列准确性超过99.999%(QV50),这也是ONT平台测序存在同聚物偏好性错误而无法自身进行校正无法比拟的。
C.均匀的覆盖度
大多数测序系统受到覆盖偏好性的困扰,从而导致富含AT或富含GC的DNA区域、高度重复序列等难以测序。这往往会导致不完整基因组覆盖率,甚至会在最终结果中造成高达15%的基因组信息缺失。单分子实时(SMRT)测序不需要扩增步骤,可实现对整个基因组的均匀覆盖。这样就能够测序回文序列和多样性程度低的基因组区域,同时长读长测序同样能够跨越复杂区域。
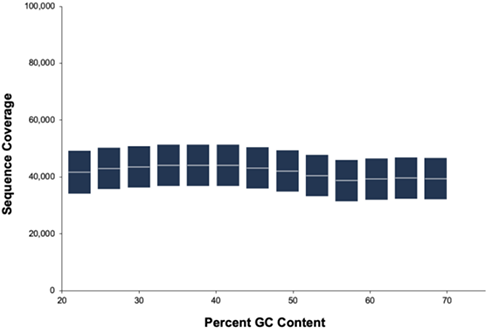
D.高精准度的长读长HiFi reads
Sequel II平台除了具有超长读长的CLR测序模式外,还可以进行兼顾读长与高精准度的HiFi reads(High fidelity reads),一般采用CCS(Circular Consensus Sequencing)模式测序。在这种测序模式下,酶读长一般大于插入片段长度,因此酶会绕着模板进行滚环测序,插入片段会被多次测序。单次测序中造成的随机测序错误,可以通过算法进行自我纠错校正,最终得到高准确度的HiFi reads。
| 关于三代测序HiFi reads你值得一看的深度好文~ |
小科普:什么是HiFi reads?
HiFi reads(High fidelity reads)是Sequel II三代测序平台推出的兼顾长读长和高准确度的测序序列,一般采用CCS(Circular Consensus Sequencing)模式测序。在这种测序模式下,酶读长一般大于插入片段长度,因此酶会绕着模板进行滚环测序,插入片段会被多次测序。单次测序中造成的随机测序错误,可以通过算法进行自我纠错校正,最终得到高准确度的HiFi reads。要在单次测序中得到更多的HiFi reads往往需要平衡测序的酶读长和插入片段的长度,插入片段太长会导致酶无法进行滚环测序,插入片段太短又牺牲了三代长读长测序的优势。因此HiFi模式测序对酶试剂和建库过程的均一性要求较高。做完科普了,小编先带大家看一下安诺近期下机的HiFi测序数据~
安诺HiFi reads数据测评
根据前期的官方经验推荐,目前HiFi文库构建的插入片段一般为8-13 kb左右。本次安诺优达构建约10 kb的HiFi文库在Sequel II平台进行测序。原始下机数据单cell产出268 Gb数据,其中酶平均读长51 kb,酶读长N50 124 kb,subreads平均读长11 kb,subreads N50 13 kb。
下机数据产出统计表

进一步利用官方软件调取CCS,设置最小pass数为3,经过调取获得CCS总数据量为22.43 Gb,CCS 序列数目为172.5万条,平均长度13 kb。与下机总数据量相比,目前CCS reads的得率约为8%,并且能够兼顾reads的读长,达到平均13 kb左右,数据质量相当不错!
CCS数据产出统计表

小编对我们拿到的HiFi reads进行进一步的质量评估,发现大部分HiFi reads的准确度都在0.95以上,其中约35%的reads(pass≥10)质量值达到QV30(99.9%),这样高质量的reads非常有助于研究者开展下游深入的研究。
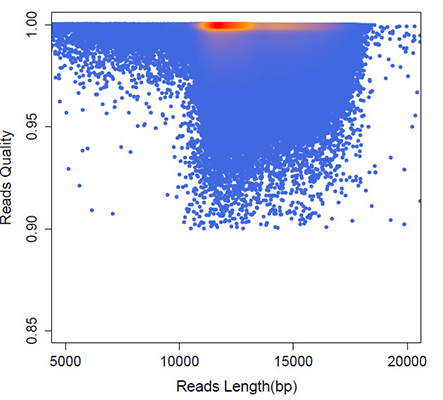
CCS质量分布图
HiFi reads有哪些用处?
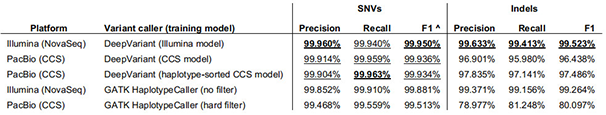
同时兼顾长读长和高准确度的HiFi reads究竟有何用处呢?小编先带大家看一篇今年发表在BioRxiv上的题为“Highly-accurate long-read sequencing improves variant detection and assembly of a human genome”的文章。在这篇文章里研究者利用约30X的CCS reads组装人基因组,通过FALCON、 Canu3和 wtdbg2等不同软件进行组装,contig N50达到15.43-28.95 Mb。从组装连续性来看,CCS reads能够做到与传统的CLR reads组装相当的结果,重要的是基因组碱基准确度得到了明显提升,基因组组装消耗的计算资源和时间大幅下降[1]。进一步利用CCS reads进行SNP、InDel等变异检测,发现CCS reads在小的变异检出率和准确度上都有显著提升,数据结果与30X的Illumina数据分析结果基本接近。
文章中CCS reads进行SNV和InDel calling统计表[1]
综上可以看出,HiFi reads无论在基因组全变异检测(SNV、InDel、SV)还是基因组de novo领域都有非常大的应用价值。目前唯一的限制因素是要获得足够的HiFi reads,测序成本的投入是比较昂贵的,但小编认为排除纯测序成本的考量,从组装计算资源节省和项目时间缩短的角度来看,HiFi reads未尝不是更好的选择。对于基因组重复序列较多的复杂基因组,目前市场上传统长读长测序准确度不高的特点给组装造成了一定的困难,高准确度的HiFi reads未来可能是一个更好的解决方案。而对于昆虫、中草药、藻类等重复序列较高、基因组较小的物种(<700 Mb),目前利用一个8 M SMRT Cell 产出的数据量基本足以支持CCS组装,性价比更高。安诺基因目前已经搭建了完善的HiFi文库建库流程和基于CCS reads组装的生信流程,期待与大家合作!
参考文献::
[1] Wenger, Pelusol, et al. Highly-accurate long-read sequencing improves variant detection and assembly of a human genome[J]. BioRxiv, 2019.
|
PacBio SMRT 测序,以其超长读长、无GC偏好性、基因组覆盖均匀等特点,在结构变异检测、基因组重测序、基因组de novo以及全长转录组测序中应用广泛。并且三代测序平台助力高水平文章发表层出不穷,常发表于Nature、Science、Nature Genetics等各大知名学术期刊,为医学和生命科学研究提供高质量数据来源。
(1)人基因组重测序——结构变异检测
结构变异,包括倒位、缺失、重复和易位,是大多数癌症基因组的标志。结构变异的发现及其对基因结构和表达的影响大大促进了我们对肿瘤和疾病发生的认识。然而,利用二代外显子和全基因组测序鉴定基因组中的结构变异仍具有挑战性。作为前沿热点技术PacBio SMRT低深度人重测序是研究结构变异的利器。
|
|
三代+Hi-C强强联合,打造医学多组学精准解决方案
在人基因组相关的疾病研究中,通过各种手段鉴定获取基因组变异信息往往是研究工作中最基础也最重要的一步。目前最常用的寻找变异信息的技术莫过于外显子测序和二代全基因组重测序,大量的变异信息通过测序被获取解读,推动了医学精准化研究的进程。
结构变异,包括倒位、缺失、重复和易位,是大多数癌症基因组的标志。结构变异的发现及其对基因结构和表达的影响大大促进了我们对肿瘤和疾病发生的认识。然而,目前利用二代外显子和全基因组测序鉴定基因组中的结构变异仍具有挑战性。作为前沿热点技术PacBio SMRT低深度人重测序和Hi-C技术都是研究结构变异的利器。Dixon JR等研究也发现在癌症基因组中存在很多因结构变异而导致的三维基因组结构改变的案例[1],而这些结构变异很可能在肿瘤发生的基因错误表达调控中起到关键作用。
|
|
|
如何利用多组学技术综合阐释肿瘤或疾病发生过程中基因异常表达调控的分子机制,安诺基因生信团队研发计算流程通过将三代人基因组重测序、Hi-C测序和转录组测序的数据相结合,分析癌症或疾病基因组中的各种结构变异模式,以及结构变异对空间三维结构的影响和对基因表达的调控作用。
整体研究思路
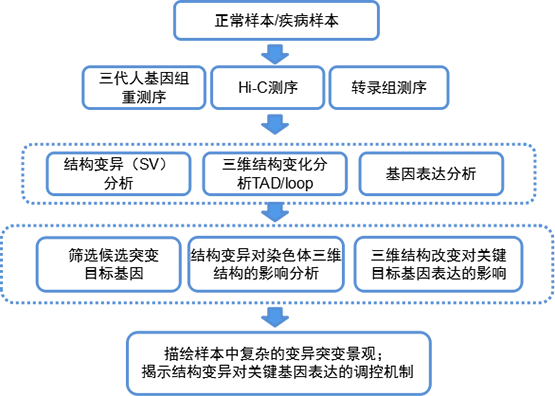
三代人重+Hi-C+转录组多组学研究思路图
部分分析 结果展示
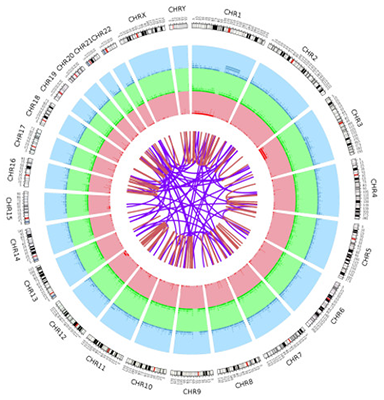
变异信息全局总览图
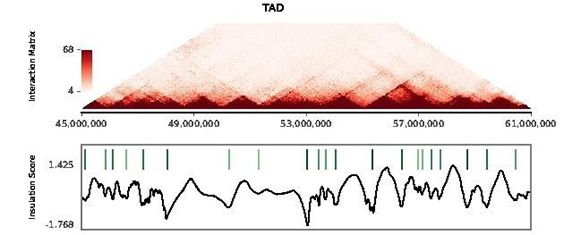
TAD总览图
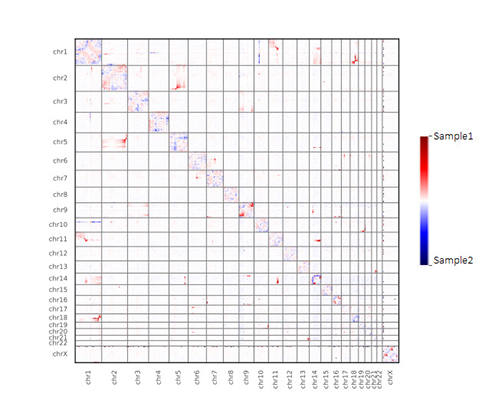
两样本差异矩阵全局互作热图
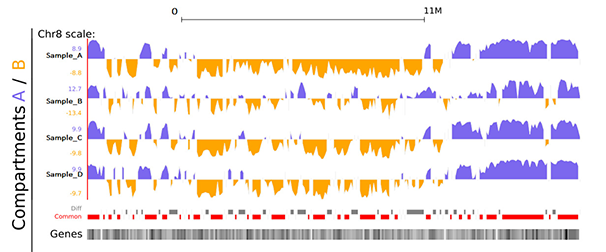
样本间差异A/B compartment分析图
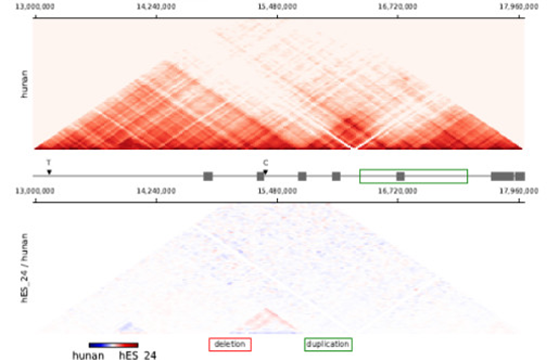
样本间差异TAD分析图
以上是基于三代人重、Hi-C、转录组测序的医学三组学研究方案,除了以上组学的联合分析,安诺三代医学多组学方案,还支持包含全基因组甲基化、ChIP-seq、蛋白质组在内的整体多组学私人定制化研究方案。
参考文献:
[1] Dixon JR, Jie X, Vishnu D, et al. Integrative detection and analysis of structural variation in cancer genomes[J]. Nature Genetics, 2018.
(2)基因组de novo中的应用——染色体水平同源多倍体单体型基因组
|
二代测序用于组装存在读长短、难于跨越基因组重复序列等局限,制约了组装结果的连续性,而三代PacBio Sequel II平台CLR模式平均读长可达20kb以上,因此在进行基因组组装中具有明显优势,尤其是组装一些同源多倍体或高重复序列的基因组。2019年8月5日,福建农林大学基因组中心张兴坦副教授和唐海宝教授研究组通过三代测序平台PacBio(113X)以及Hi-C(100X)数据,利用ALLHi-C算法解决了同源多倍体基因组组装的技术难题,成功完成了同源四倍体和同源八倍体甘蔗染色体组装,安诺基因作为合作单位有幸参与了该项目的研究工作。
|
|
基于ALLHiC算法组装染色体水平同源多倍体单体型基因组
2019年8月5日,福建农林大学基因组中心张兴坦副教授和唐海宝教授研究组在Nature Plants杂志在线发表题为“Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data”的研究论文,该研究利用ALLHi-C算法解决了同源多倍体基因组组装的技术难题,成功完成了同源四倍体和同源八倍体甘蔗染色体组装,安诺基因作为合作单位有幸参与了该项目的研究工作。
|
|
|
研究背景
同源多倍体在植物中较为常见,一般是由于相同的两套或多套基因组经过加倍形成的,有重要的遗传育种和农业生产价值。然而除了已发表的甘蔗割手密基因组[1]外,染色体级别的同源多倍体基因组很少被破译出来。目前Hi-C技术越来越多的应用于辅助染色体水平二倍体基因组组装,但是对于同源多倍体和近期加倍的异源多倍体来说,其同源染色体之间的Hi-C交联信号会将序列相似的等位基因片段连接在一起,导致同源染色体被错误地连接到一起,形成大量嵌合的组装,所以其组装仍存在较大困难。本研究中研究者利用ALLHi-C算法突破了同源多倍体染色体组装的技术困境,取得了开拓性的进展。
材料选择
同源四倍体甘蔗AP85-441,同源八倍体甘蔗Molokai-6081
研究结果
ALLHiC算法和验证数据集概述
ALLHiC算法包括pruning,partition,rescue,optimization,building5个步骤,通过修剪同源染色体之间的交联信号,将等位基因和同源序列分隔在各自的单倍型内独立组装,从而减少了大量拼接错误,通过优化算法改进了contig的排序和定向,尤其是连续性较低的contig,成功解决了染色体水平同源多倍体组装困难的问题。文章通过“合成的”或者真实的基因组数据集验证了ALLHiC算法的可行性。通过将两个栽培稻亚种Oryza sativa spp.japonica和O.sativa indica组合构建出合成基因组,再将染色体分成不同的contig组合,进行了一系列模拟,与真实基因组序列对比,有效验证了ALLHiC算法的适用性。影响ALLHiC组装scaffold的因素包括contig N50,嵌合区和冲突区占比以及序列多样性。
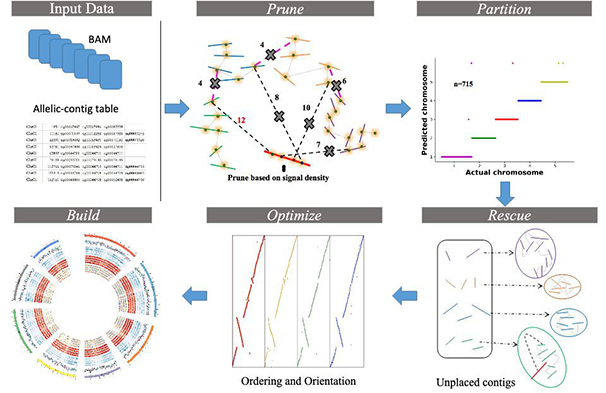
Fig.1 ALLHiC算法流程和功能模块[2]
应用ALLHiC算法组装同源四倍体甘蔗基因组
甘蔗AP85-441是通过Saccharum spontaneum花药培育的同源四倍体(1n = 4x = 32),其基因组已发表,共32条染色体,8套同源染色体,每套染色体有4个单倍型,组装出基因组大小为3.13 Gb,congtig N50为45 kb,文章以Chr4同源染色体组为例说明了ALLHiC算法在组装染色体水平同源四倍体甘蔗基因组中的应用。
基于BLAST方法将预测的甘蔗AP85蛋白与高粱基因组比对来鉴定其等位基因contig,共8,107个注释蛋白被鉴定为2,993个等位基因contig,4,167个非冗余contig,共11,292,703条in situ Hi-C reads比对到等位基因contig上。通过修剪同源染色体Hi-C交联信号,将等位基因contig分区,优化算法进行准确排序和定向后成功组装出了相应的scaffolds。文章验证了所有scaffolds都保持了高粱和甘蔗基因组之间的高共线性,说明二者分歧时间较短,与先前遗传图谱研究结果一致。最终90.93%(3,789 / 4,167)的等位基因contig聚类组装成一组4个单倍型的同源染色体,占总contig长度的94.47%(183.85 / 194.61 Mb)。Hi-C热图显示每个同源染色体与其他染色体间没有强相互作用。以上证明运用ALLHiC算法能成功组装出染色体水平同源四倍体甘蔗基因组。
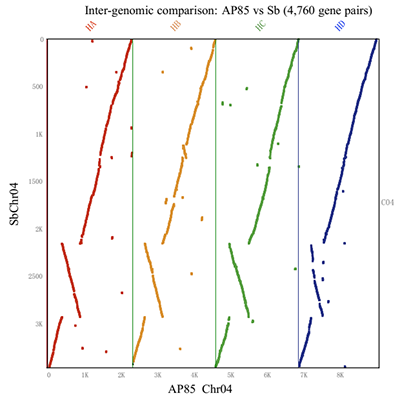
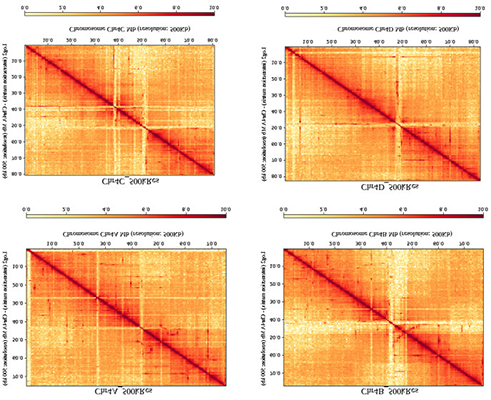
Fig.2 ALLHiC算法组装同源四倍体甘蔗基因组S. spontaneum AP85-441的scaffolds[2]
应用ALLHiC算法组装同源八倍体甘蔗基因组甘蔗Molokai-6081是Saccharum robustum(2n=60-170)的同源八倍体,Molokai基因组内的某些染色体组可能会出现非整倍性,以Chr5同源染色体组为例,运用ALLHiC算法,生成了16个super-scaffolds,进一步研究它们之间的信号密度,重新聚类成9个super-groups,优化后进行排序定向,最终共12,077个contig,98.65%的序列锚定在Chr5,染色体长度为46-98Mb,证明ALLHiC算法可应用于组装染色体水平同源八倍体甘蔗基因组。
ALLHiC算法在异源多倍体和高杂合二倍体基因组scaffold构建中的应用
异源四倍体栽培花生(Arachis hypogaea L.)基因组由两个亚基因组组成,可能源于二倍体Arachis duranensis(AA)和Arachis ipaensis(BB)杂交形成。这两个亚基因组的分歧时间非常短。100X PacBio测序数据组装花生基因组大小为2.54 Gb,contig N50为1.51 Mb,将100X Hi-C reads比对到花生基因组contig,使用ALLHiC算法组装出scaffolds与公布的花生基因组一致性高达83.05%。除此之外,将最近发表的水稻Nipponbare和93-11构建成高杂合二倍体基因组,也显示出ALLHiC算法广泛的适用性。
文章总结
ALLHiC算法一方面通过修剪Hi-C平行信号和弱信号进行等位基因分型,减少了同源染色体间的嵌合连接,另一方面通过遗传算法随机优化,极大地提高了短序列的排序和定向准确性。ALLHiC算法使多种重要多倍体基因组直接从头组装成为可能,还可用于修复已公布的多倍体物种基因组组装序列中的错误。ALLHiC算法除了适用于同源多倍体染色体组装外,同样适用于不同复杂度的基因组,包括简单的二倍体基因组、高杂合基因组和异源多倍体基因组,极大地推动了基因组领域的研究发展。
参考文献
[1] Zhang J, Zhang X, Tang H, et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L[J]. Nature Genetics. 2018.
[2] Zhang X, Zhang S, Zhao Q, et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data[J]. Nature Plants. 2019.
(3)Iso-Seq测序——研究肝细胞癌HCC可变剪切变体
|
Iso-seq技术无需拼接可直接获得全长转录本,克服了二代转录组测序存在的长度短、需拼接等困难,因此在可变剪切(AS)、可变聚腺苷酸化(APA)、融合基因、LncRNA等的预测及分析方面更具优势。这里小编挑选了一篇经典医学案例来解析iso-seq在研究肝细胞癌HCC可变剪切变体方面的应用~的轻狂
|
|
肿瘤特异可变剪切变体经典案例分享
近年来iso-seq在医学方面也文章频出,如研究人骨髓细胞亚群的isoform[1]、KIR受体基因的大量可变剪切[2,3]、肿瘤细胞剪切因子NOVA1[4]、乳腺癌细胞复杂基因结构[5,6]等。今天小编挑选了一篇经典医学案例来聊聊iso-seq在研究肝细胞癌HCC可变剪切变体方面的应用~的轻狂
|
|
|
文章题目:长读长测序鉴定肝细胞癌的可变剪切变体和肿瘤特异性isoform[7]
发表时间:2019年1 月
发表期刊:Hepatology(IF:14.079)
样本选择:
三代iso-seq:HCC患者细胞和MIHA细胞系;
二代RNA-seq:HCC细胞、MIHA细胞系、HCC癌及癌旁组织、正常肝组织
实验设计
实验结果
01可变剪切(AS)变体整体分析
研究中对8例来源于患者的HCC细胞和MIHA细胞系进行SMRT测序,共获得8,990条全长转录本,其中362条是已知位点的新isoform(图A)。与已注释isoform相比,新isoform的平均长度较短;分析发现剪切因子的异常调节与RI和SE类型AS事件的产生有关,且两种事件的发生呈负相关(图B)。对新isoform进行GO富集分析,发现它们在RNA结合、酶结合及调节、转录活性等方面显著富集(图C)。这些通路中基因的剪切变体可能通过其他机制调控HCC细胞中的癌症相关通路。
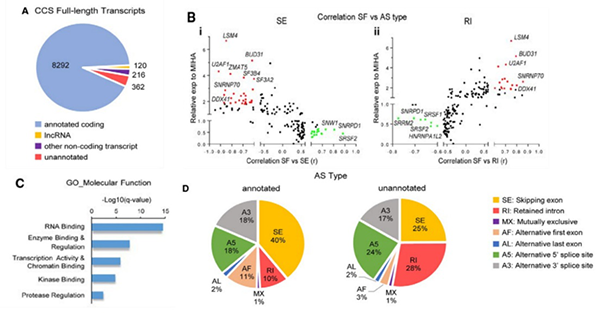
可变剪切分析[7]
02 肿瘤特异性isoform分析
研究中将正常肝组织、MIHA和HCC细胞中的转录本进行对比,共获得2,057条HCC细胞特有的转录本(图A)。51.7%的基因功能在酶结合及调节、受体结合和转录调节等方面富集(图B)。与成对的癌旁组织相比,在约50%的HCC肿瘤样本中DEK和ADRM1变体的表达上调超过10倍,少数样本甚至上调超过100倍;同样地,SRSF3、ROR1和VDR变体在HCC中也有较明显的表达上调(图DE)。这些结果表明上述的AS变体表达水平或许可以暗示肝癌发展进程。
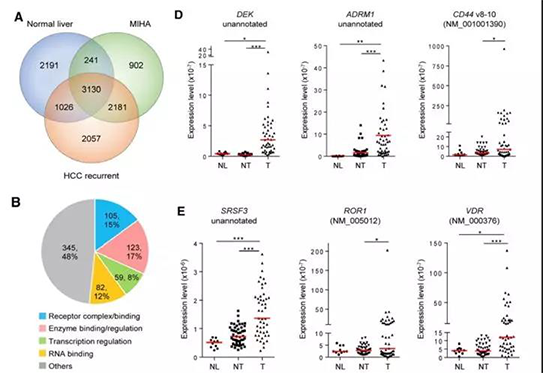
肿瘤特异性isoform分析[7]
03 ARHGEF2变体的生物学和临床显著性
ARHGEF2在激活HCC的致癌RhoA信号通路中起重要的作用。为探究ARHGEF2的v1和v3变体(图A)在肝癌患者中的关联性,作者在10例正常组织、87例肿瘤及其成对的癌旁组织中对它们的表达进行了定量检测:v1和v3在正常组织中几乎不表达,而在癌旁组织中显著上调;与癌旁组织相比,肿瘤组织中v1和v3出现平均3-4倍的上调(图D),并表现出很强的线性相关性(图E)。这表明v1和v3变体在肿瘤组织中共表达,且可以暗示肝癌的早期变化。
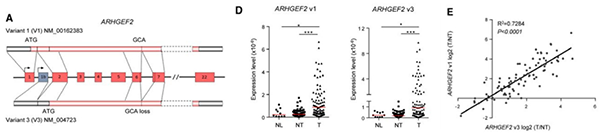
ARHGEF2变体在HCC患者中的表达分析[7]
之后作者将ARHGEF2变体的表达与肝细胞癌的临床病理进行相关性分析。研究发现v1和v3的高表达与微血管侵犯显著相关,昭示着肝癌的传播和转移(图A);生存分析中,v1和v3的表达量越高,对应的患者的存活率越低(图C)。此外,通过将变体表达量与肿瘤等级相关联,发现v1变体的高表达与2级和3级肿瘤也高度相关(图B)。
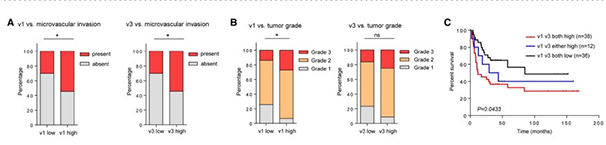
ARHGEF2变体的临床病理分析[7]
为探究AS变体是否有特异的生物学功能,研究者构建了ARHGEF2敲除细胞系HKCI-8和Hep3B,并在MIHA细胞系和敲除细胞系中将v1和v3变体过表达。数据显示,虽然许多癌症相关基因会促进细胞生长,但v1和v3不具有影响细胞生存的能力。v1和v3的表达会诱导EMT(上皮向间叶细胞转化)过程的发生,使细胞获得高迁移和入侵能力,且v3的效果更显著(图BC);v1的过表达则能提高RhoA的活性,促进其信号通路在细胞运动中的作用。此外,通过分析细胞干性标志物的表达水平,作者还发现v1和v3的表达能增强细胞的肿瘤干性(图DEF)。
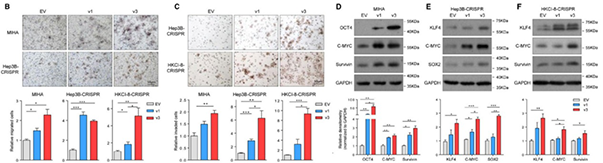
AS变体特异性生物学功能分析[7]
综上,研究中利用全长转录组测序鉴定了HCC细胞新的和特有的isoform,并对它们的表达模式和特异性生物学功能进行了分析。研究结果强调了三代测序技术在鉴定可变剪切事件上的优势,且AS变体可能作为肿瘤治疗的新biomarker或分子靶标,为HCC的治疗和预后提供有力支持。
参考文献:
[1]Deslattes Mays, A., Schmidt, M., Graham, G., et al. Single-Molecule Real-Time (SMRT) full-length RNA-sequencing reveals novel and distinct mRNA isoforms in human bone marrow cell subpopulations[J]. Genes, 2019.
[2] Bruijnesteijn Jesse., van der Wiel Marit K H., de Groot Nanine., Otting Nel., et al. Extensive alternative splicing of KIR transcripts[J]. Front Immunol, 2018.
[3] Bruijnesteijn, J., Van, M. D. W., Swelsen, W., et al. Human and rhesus macaque kir haplotypes defined by their transcriptomes[J]. Journal of Immunology, 2018.
[4] Sayed Mohammed E., Yuan Laura., Robin Jerome D., et al. NOVA1 directs PTBP1 to hTERT pre-mRNA and promotes telomerase activity in cancer cells[J]. Oncogene,2018.
[5] Maria, N., Sara, G., Karen, N., et al. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line[J]. Genome Research, 2018.
[6] Anvar, S. Y., Allard, G., Tseng, E., et al. Full-length mRNA sequencing uncovers a wide spread coupling between transcription initiation and mRNA processing[J]. Genome biology, 2018.
[7] Chen Hui., GaoFeng., He Mian., et al. Long-read RNA sequencing identifies alternative splice variants in hepatocellular carcinoma and tumor-specific isoforms[J]. Hepatology,2019
高质量的数据产出
1000+项目经验
领先的分析团队
优质高效的服务体系
自2017年推出三代测序服务以来,安诺优达先后引进了10台PacBio Sequel,并在今年又引入4台Sequel II测序仪,在成为全球首批PacBio SequelⅡ测序服务运营商后,于2019年7月获得PacBio官方认证资质,我们将继续秉承客户至上的服务理念为合作伙伴提供更优质、更快速的三代测序服务。

官方认证资质
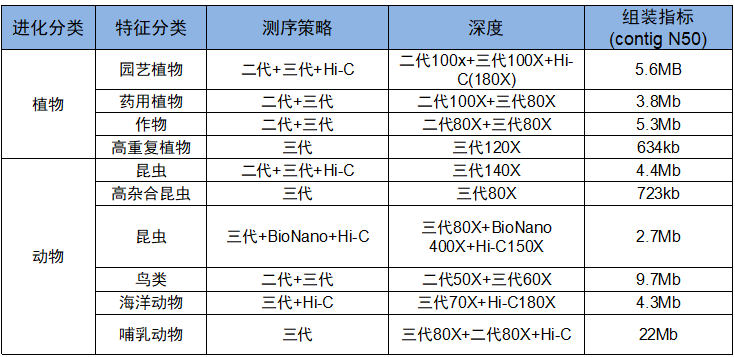
上千例项目数据显示,安诺PacBio平台运行稳定,数据产出可观,产品服务类型涵盖三代基因组组装、人重测序、动植物重测序、全长转录组测序等;具有丰富的项目经验,累计完成三代项目超800+,其中组装经验涉及中草药、林木、农作物、海洋生物、哺乳动物、昆虫和人等,组装结果一致性评估可达90%以上,Hi-C辅助组装挂载率可达95%以上,并发表多篇合作文章。全长转录组项目物种经验涵盖林木、作物、中草药、哺乳动物、水生生物和昆虫等,总数达150+,目前已合作项目300+,建库成功率超98%。以下是一组Sequel II实测数据及基因组de novo部分组装结果统计。
(1)安诺Sequel II实测数据展示:
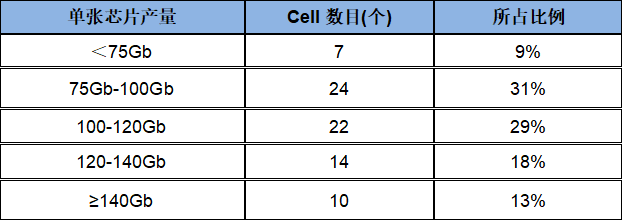
对Sequel II平台CLR模式77张SMRT cell测试数据产量及平均读长进行统计,其中数据产量结果分布显示超过90%的cell 数据产出超过75Gb,60%的 cell产出超过100Gb,最高产量达到153Gb,平均产出107Gb。
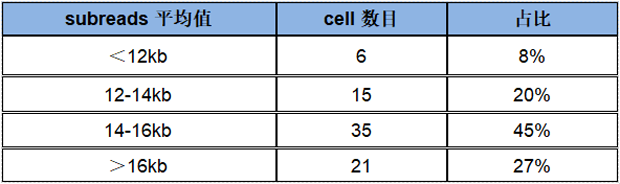
单张cell平均读长统计显示超过92%的cell subreads 平均读长在12kb以上,其中72%的cell subreads 平均读长超过14kb。
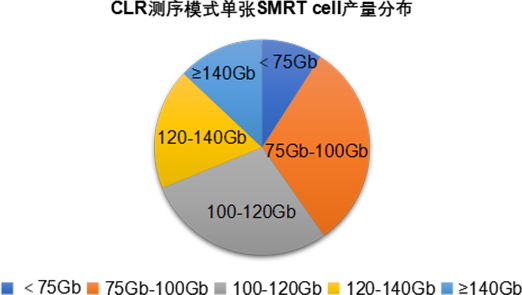
Sequel II单张SMRT cell产量统计表
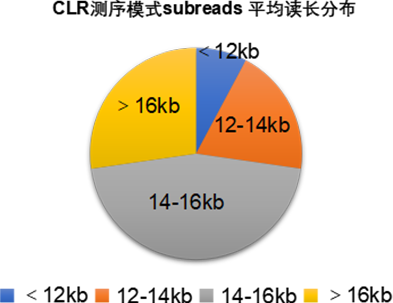
Sequel II单张SMRT cell平均读长统计表
(2)高质量的基因组组装:
基因组de novo 组装指标
|
