
-

生物通官微
陪你抓住生命科技
跳动的脉搏
基于Iso-seq数据的蛋白质组学分析新方法
【字体: 大 中 小 】 时间:2022年09月02日 来源:
编辑推荐:
今天向大家介绍由来自美国威斯康星大学麦迪逊分校的学者本月初发表于Genome Biology的一项开创性的蛋白isoform鉴定方法。
全长转录组测序(Iso-Seq)是基于PacBio单分子实时测序技术,凭借超长读长的优势,无需打断RNA分子,直接对反转录的全长cDNA测序,即可得到从5’末端到3’PolyA尾的高质量全长转录本序列,从而对同源异构体(isoform)、可变剪接(AS)、融合基因、同源基因、超家族基因、等位基因表达等进行精确分析的一项技术。
基因由于AS或可变启动子最终形成不同的蛋白质产物,这些源自同一基因的蛋白isoforms在细胞中表现出不同的稳定性、分子结合能力和功能效应。研究表明,许多蛋白isoform与神经退行性病变、癌症等多种疾病有关。通过转录测序估计,可能存在超过30万个人类蛋白亚型。然而,很少有实验方法能够轻易地在isoform分辨率下检测到蛋白质,也不知道转录异构体的复杂性能够多大程度上传递到蛋白质组。
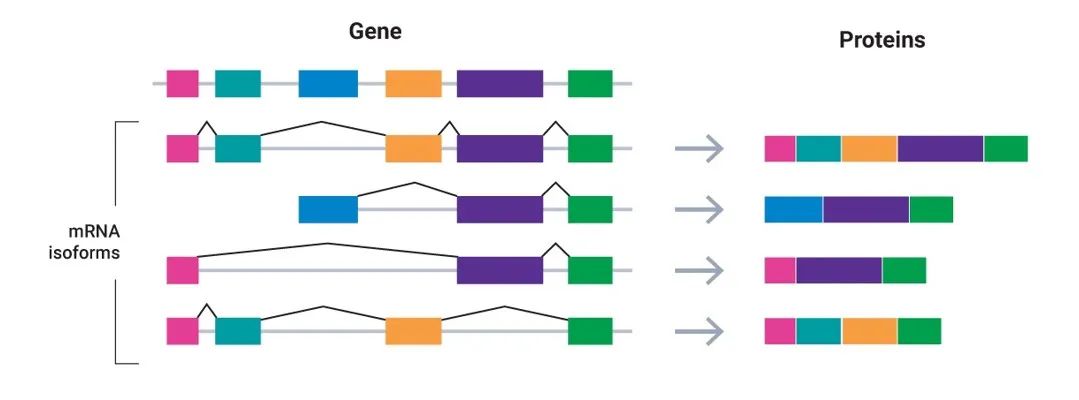
可变剪接是产生人体蛋白质多样性的基础
蛋白质谱(MS)分析方法得到的肽段数据比对参考蛋白质数据库来推断蛋白isoform是很困难的,因为蛋白亚型之间存在大量的肽段共享,并且参考蛋白质数据库广泛地代表了一个生物体的蛋白质组,无法区分不同组织、发育和疾病状态以及个体之间的蛋白质组差异,也不能发现新的蛋白isoform。
今天向大家介绍由来自美国威斯康星大学麦迪逊分校的学者本月初发表于Genome Biology的一项开创性的蛋白isoform鉴定方法。该研究整合基于MS蛋白质组学数据和PacBio Iso-seq数据,开发了一个用于蛋白isoforms鉴定的长读长(Long read,LR)蛋白质基因组学pipeline,从而检测到了以前基于MS难以鉴定的蛋白isoforms。

实验使用Jurkat T淋巴细胞系,具体工作流程如下:
✓PacBio测序分析,揭示高质量的全长转录序列
✓预测转录本的开放阅读框(ORF)
✓用SQANT1 Protein工具执行蛋白isoform分类
✓使用PacBio数据和GENECODE参考isoform数据生成样品特异性的全长蛋白质数据库
✓开发一种新的蛋白质推断算法,通过PacBio数据获得的转录本丰度值增加了蛋白isoform识别的数量
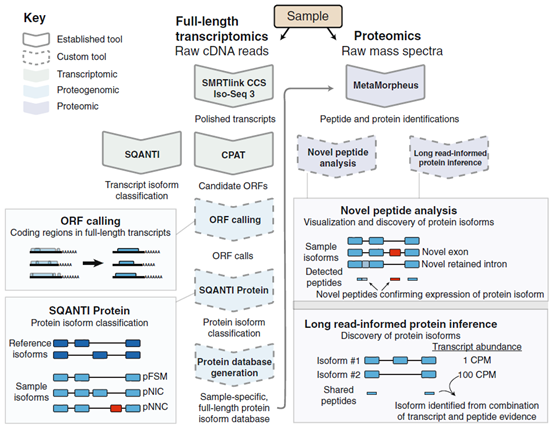
工作流程示意图
1、LR RNA-seq 揭示GENCODE数据中不存在的转录isoforms
作者在PacBio Sequel II平台进行Iso-seq获得HiFi数据,经过除去测序接头、除去polyA尾、isoform聚类、参考基因组(hg38)比对等处理,然后使用SQANT1工具比较LR RNA-seq isoforms与GENCODE数据库的参考转录本(v35)发现:
❖有43865条转录本与GENEODE完全匹配――full splice matches,FSMs
❖有43075条转录本是新发现的,但为已知的剪接位点重新组合而来――novel in catalog,NICs
❖有32416条转录本含有以前未发现的剪接位点或新的外显子――novel not in catalog,NNCs
2、转录本开放阅读框(ORF)预测
作者开发了一个编码潜能评估工具(Coding-Potential Assessment Tool,CPAT),该工具能对每个转录isoform对应的ORF进行评分。结果发现91%的转录isoforms对应着唯一的高置信度ORF(CAPT score>0.9),剩下9%的转录本存在多个高分ORF,不能明确唯一的ORF。所以后续作者又在CPAT评分的基础上加上了ATG起始密码子的GENCODE注释状态和起始密码子相对于5 '端位置两个指标,实现了所有转录本的唯一ORF预测。一些不同的转录isoforms可能具有相同的编码区,产生相同的理论蛋白质亚型产物。
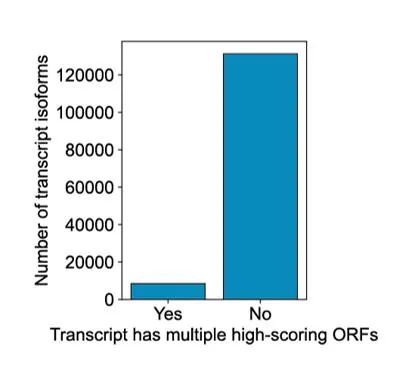
具有一个或多个高分ORF的转录isoform计数(CPAT分数>0.9)
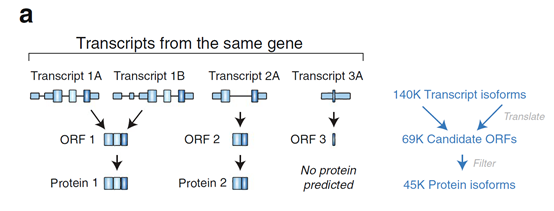
根据全长转录本预测ORF和蛋白序列
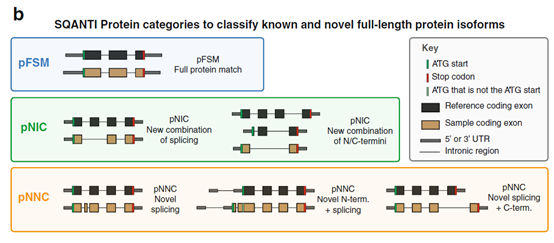
3、SQANTI Protein――全长蛋白质isoforms的新分类方案
根据ORF预测结果导出基因的蛋白isoforms模型,发现许多基因可以同时表达多种蛋白质亚型。蛋白isoform分类考虑的是全长蛋白质序列,只能通过LR RNA-seq检测,这是只关注“局部”的技术例如的短读长RNA-seq和芯片技术所不能达到的。
为了系统地描述这些全长蛋白亚型,作者创建了一个新的蛋白亚型分类方案――SQANTI Protein,以描述预测的蛋白isoform与GENCODE中注释的蛋白isoform之间的关系。该方案基本遵循SQANTI中的分类原则,也将蛋白isoforms分为:
❖16331(24%)与GENCODE中注释的蛋白完全匹配――pFSM
❖7642 (11%)的序列元件在参考文献中有注释,但元件的组合是新的――pNIC
❖21095 (30%)至少含有一个新元件――pNNC
新预测的蛋白isoforms构成了数据库的重要部分。其中75%基因至少有一个pNIC或pNNC;三分之一的基因对应的丰度最高的蛋白isoform与GENECODE 中的参考isoform不对应;且42.5%(1215)的蛋白isoforms是之前未发现过的,这进一步说明了构建样品特异性的蛋白数据库的重要性。于是作者接下来将SQANTI Protein注释的、覆盖10348个基因的45068个蛋白亚型(包括pFSM、pNIC和pNNC)生成样品特异的数据库。
4、生成高置信度的PacBio衍生蛋白数据库
作者对PacBio数据集设置过滤掉条件,过滤掉极端长度(<1kb或>4kb)、低丰度(例如CPM<3)和没有3 ' polyA覆盖的数据,生成具有可信的完整蛋白isofom模型候选基因,最后有6653个基因在该高置信(High confidence ,HC)区间。然后使用GENCODE数据库来补充其他未覆盖到基因的蛋白质数据,生成一个PacBio-derived(6653个基因对应35119种蛋白质)和GENCODE(13276个基因对应48413种蛋白质)的混合数据库――PacBio-Hybrid数据库,作为下游蛋白质组分析的参考数据库。
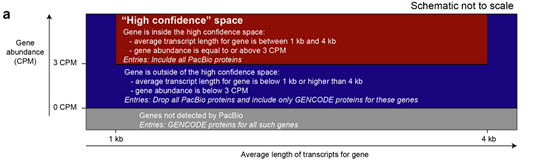
高置信度PacBio-derived数据生成
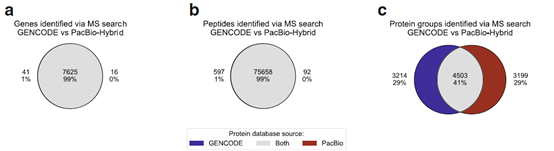
5、使用PacBio衍生的蛋白质数据库对基于MS的蛋白质组进行分析
作者使用同一来源的Jurkat细胞系进行蛋白质谱,分别根据PacBio-Hybrid数据库与GENCODE参考数据库进行蛋白组学分析,两个数据库99%的基因鉴定和99%的肽是匹配的,这表明两个数据库接下来用于蛋白质推断的肽集几乎相同。但从下图看出,二者只有41%(4503)的蛋白isoforms是相同的。因为在多肽印射回蛋白isoform的过程中,两个数据库中isoforms组成差异和共享多肽分配到其来源的蛋白isoform的不确定性导致二者蛋白质鉴定的差异。而PacBio来源的样本特异性数据库给没有足够特异性肽段支持的isoforms提供了转录证据,提升了蛋白质亚型鉴定的准确性和精密度。
在PacBio-Hybrid数据库中进一步检索,发现了GENCODE和UniProt参考数据库中都没有的14个新肽序列,这些新肽序列由上游ATG起始位点的使用、内含子保留和新外显子翻译事件产生。
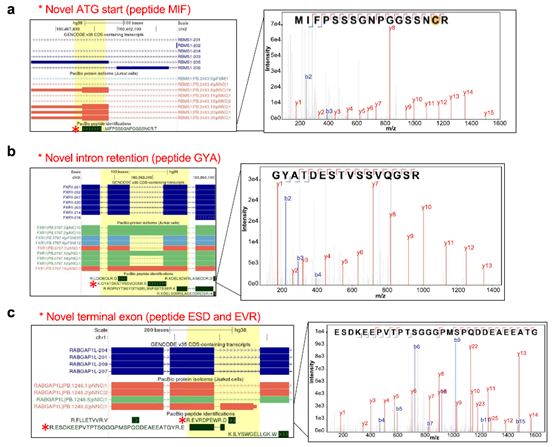
新发现的多肽和全长蛋白isoforms
最后,作者使用每种转录isoform的测序reads数来评估样品中每个isoform的相对丰度,将转录丰度值整合到蛋白质推断过程中,开发了一种启发式的蛋白质推理算法,称为Rescue& Resolve (R&R),该算法使源于PacBio数据分析到的蛋白isoforms数量又增加了355个,并且将原本1434个不明确的蛋白isoform groups分解为单个蛋白isoform进行鉴定,强调了其中295个isoform groups的存在。
总之,这篇文章介绍了一种新的蛋白质亚型的分类方案,即从LR RNA-seq衍生的样本特异性蛋白isoform模型来增强蛋白isoform的鉴定,检测迄今无法用MS分析的蛋白质亚型。将LR RNA-seq与蛋白组学相结合,改进人类蛋白isoform多样性表征,为未来长读长蛋白基因组学的发展及其应用于基础和转化研究奠定了坚实的基础。
文献链接:Enhanced protein isoform characterization through long-read proteogenomics
基因有限公司作为PacBio公司在中国区的独家代理商,自2011年以来将PacBio第三代单分子实时测序技术引入国内,一直为国内用户提供专业的三代测序系统的安装培训,技术支持,应用培训与售后维护工作,赢得客户的一致好评与信任。基因有限公司将一如既往的支持越来越多的PacBio用户。




