
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
基于最大熵重加权方法整合分子动力学模拟与实验数据解析固有无序蛋白原子分辨率构象系综
【字体: 大 中 小 】 时间:2025年10月12日 来源:Nature Communications 15.7
编辑推荐:
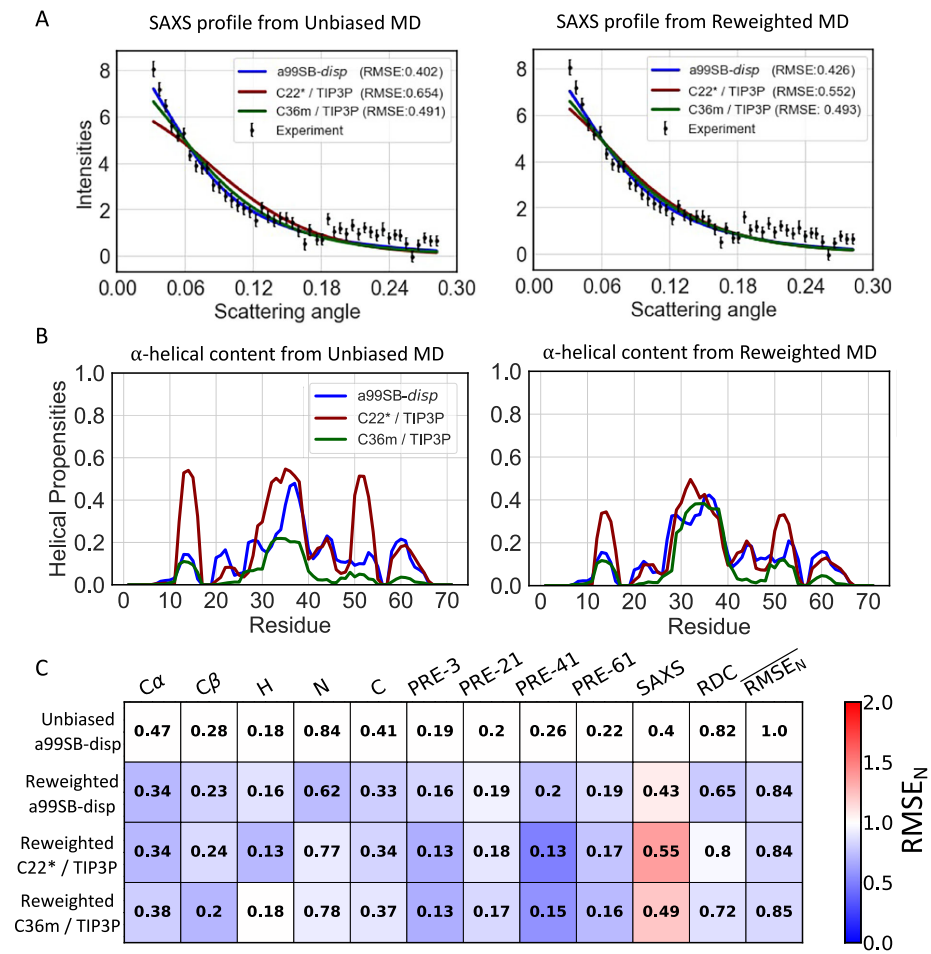
本研究针对固有无序蛋白(IDPs)原子分辨率构象系综测定难题,开发了一种全自动最大熵重加权方法,成功整合全原子分子动力学(MD)模拟与核磁共振(NMR)、小角X射线散射(SAXS)实验数据。研究证明该方法可在不同力场初始构象分布合理的情况下,获得高度一致的力场无关性构象系综,为IDP精准结构表征及药物靶点开发提供了重要技术支撑。



 生物通微信公众号
生物通微信公众号
知名企业招聘

