
-

…ķőÔÕ®ĻŔőĘ
Ň„ń„◊•◊°…ķ√ŁŅ∆ľľ
ŐÝ∂ĮĶń¬Ų≤ę
…ķőÔÕ®ĻŔőĘ
Ň„ń„◊•◊°…ķ√ŁŅ∆ľľ
ŐÝ∂ĮĶń¬Ų≤ę
◊Ř Ų£ļŐķňņÕŲ£ļ∑÷◊”Ľķ÷∆°Ę≤°ņŪ…ķņŪ—ßľį∆š‘ŕ∂ýŅ∆∑ő≤Ņľ≤≤°÷–Ķń◊ų”√
°ĺ◊÷ŐŚ£ļ īů ÷– –° °Ņ Īľš£ļ2025ńÍ10‘¬29»’ ņī‘ī£ļCell Biology and Toxicology 5.9
Īŗľ≠Õ∆ľŲ£ļ
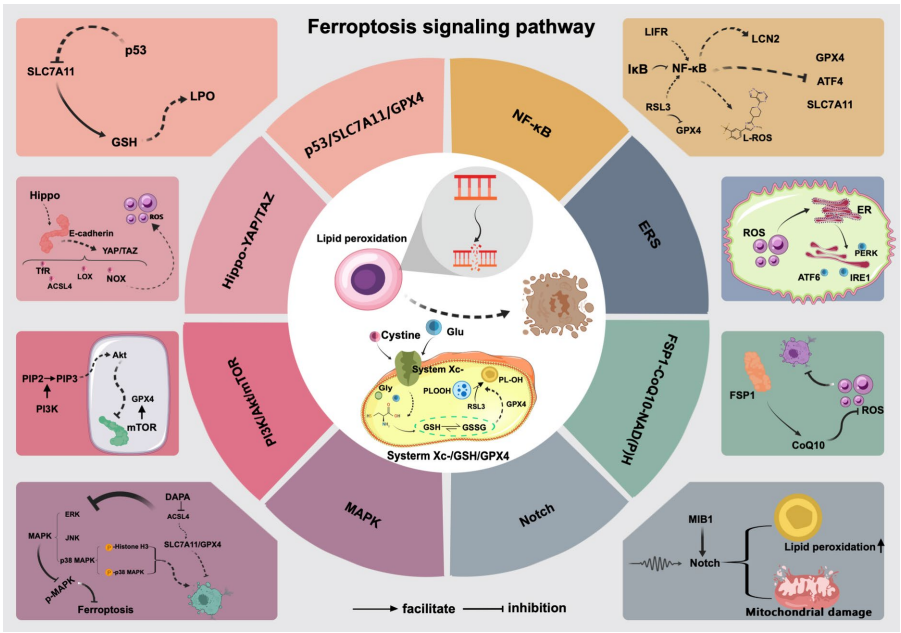
°°°°Īĺ◊Ř ŲŌĶÕ≥≤Ż ŲŃňŐķňņÕŲ£®Ferroptosis£©’‚“ĽŐķ“ņņĶ–‘°Ę÷¨÷ Ļż—űĽĮ£®LPO£©«ż∂ĮĶń≥Ő–Ú–‘ŌłįŻňņÕŲ–ő Ĺ°£őń’¬…Ó»ŽŐĹŐ÷Ńň∆šļň–ń∑÷◊”Ľķ÷∆£¨įŁņ®Őķīķ–Ľ°ĘĽÓ–‘—ű£®ROS£©…ķ≥…°ĘĻ»Ž◊ł Žń£®GSH£©/Ļ»Ž◊ł ŽńĻż—űĽĮőÔ√ł4£®GPX4£©÷Š°Ęp53/SLC7A11/GPX4–ŇļŇ÷Š“‘ľįFSP1-CoQ10-NADPHÕ®¬∑Ķ»ĻōľŁĶųŅōÕݬÁ°£÷ōĶ„∑÷őŲŃňŐķňņÕŲ‘ŕ∂ýÕĮŌÝī≠°ĘļŰőŁĶņł–»ĺ£®»ÁĹŠļň∑÷÷¶łňĺķMtB°ĘCOVID-19°ĘÕ≠¬ŐľŔĶ•įŻĺķPA£©°Ęľš÷ –‘∑ő≤°£®ILD£©°Ę∑ő∂Į¬ŲłŖ—Ļ£®PAH£©°ĘľĪ–‘∑őňū…ň/ľĪ–‘ļŰőŁĺĹ∆»◊ŘļŌ’ų£®ALI/ARDS£©°Ę÷ß∆ÝĻ‹∑ő∑Ę”ż≤ĽŃľ£®BPD£©ľįń“–‘Ōňő¨ĽĮ£®CF£©Ķ»≥£ľŻ∂ýŅ∆∑ő≤Ņľ≤≤°÷–Ķń«Ī‘ŕ≤°ņŪ…ķņŪ—ßĹ«…ę£¨≤Ę’ĻÕŻŃň“‘ŐķňņÕŲő™į–Ķ„Ķń«Ī‘ŕ÷őŃ∆≤Ŗ¬‘£¨ő™∂ýŅ∆ļŰőŁŌĶÕ≥ľ≤≤°Ķń∑ņ÷őŐŠĻ©Ńň–¬ ”Ĺ«°£


 …ķőÔÕ®őĘ–ŇĻę÷ŕļŇ
…ķőÔÕ®őĘ–ŇĻę÷ŕļŇ
÷™√Ż∆ů“Ķ’–∆ł
ĹŮ»’∂ĮŐ¨ | »ň≤Ň –≥° | –¬ľľ ű◊®ņł | ÷–ĻķŅ∆—ß»ň | ‘∆’ĻŐ® | BioHot | ‘∆Ĺ≤Ő√÷Ī≤• | ĽŠ’Ļ÷––ń | ŐōľŘ◊®ņł | ľľ űŅž—∂ | √‚∑— ‘”√
įś»®ňý”– …ķőÔÕ®
Copyright© eBiotrade.com, All Rights Reserved
Ń™ŌĶ–ŇŌš£ļ
‘ŃICPĪł09063491ļŇ
