综述:血红蛋白病的基因治疗:临床试验结果与造血干细胞及骨髓微环境生物学
《Cell Reports Medicine》:Gene therapy for hemoglobinopathies: Clinical trial results and biology of hematopoietic stem cell and the bone marrow niche
【字体:
大
中
小
】
时间:2025年11月23日
来源:Cell Reports Medicine 10.6
编辑推荐:
本综述系统梳理了血红蛋白病基因治疗领域从慢病毒基因添加到CRISPR基因编辑的临床转化突破,并前瞻性地揭示了疾病状态下造血干细胞(HSC)与骨髓(BM)微环境的生物学改变。文章深度解析了已获批疗法(如beti-cel, lovo-cel, exa-cel)的临床试验数据,同时指出无效红细胞生成、HSC功能受损及骨髓微环境缺陷是影响疗效持久性的关键因素。作者强调,未来治疗策略需结合针对HSC-BM niche互作的干预,以优化基因校正细胞的植入与功能,代表了该领域从单纯基因校正迈向综合治疗的新方向。
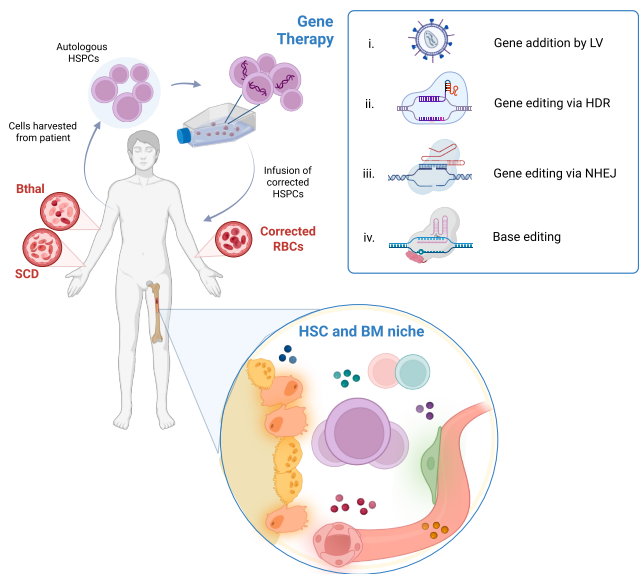
基因治疗策略:从基因添加到基因编辑
血红蛋白病,主要包括β-地中海贫血(Bthal)和镰状细胞病(SCD),是全球最常见的单基因遗传病。其根本病因在于血红蛋白(Hb)的β-珠蛋白链基因突变。在SCD中,β-珠蛋白基因的一个A>T点突变导致谷氨酸被缬氨酸取代,形成异常的血红蛋白S(HbS)。HbS在脱氧状态下会发生聚合,引发红细胞镰变、溶血、血管闭塞危象和多器官损伤。而在Bthal中,超过400种不同的β-珠蛋白基因突变导致α-珠蛋白与β-珠蛋白链比例失衡,成人血红蛋白(HbA)合成不足。其临床严重程度取决于α-珠蛋白链过剩的程度以及残余β-珠蛋白的产量。最严重的患者需要依赖长期输血和铁螯合治疗,并伴有无效红细胞生成、肝脾肿大和铁过载并发症。
针对这些疾病,基因治疗(GT)的发展经历了从基因添加到基因编辑的演进。基因添加策略主要依赖于慢病毒载体(LV)将功能正常的β-珠蛋白或具有抗镰变功能的β-样珠蛋白基因导入患者自身的造血干细胞和祖细胞(HSPCs)中。首个开创性临床试验使用表达突变β-珠蛋白T87Q(HPV569)的LV,成功使一名Bthal患者获得输血独立性,尽管在12年后的长期随访中又需要间歇性输血。该病例中,一个携带载体整合至HMGA2基因的髓系优势克隆的出现,提示了有限的移植HSC数量可能导致具有体内选择性优势的克隆扩张,但该克隆后期消退,且为良性。这一经验促使后续研究对LV-珠蛋白载体设计和CD34+细胞转导方案进行优化。
基于优化后的LV(BB305-LentiGlobin)的临床试验取得了显著成功。在关键的三期临床试验中,91%的非β0/β0基因型Bthal患者和89%的严重基因型Bthal患者在接受beti-cel治疗后实现了输血独立。同样,该策略应用于SCD(lovo-cel)也在多数患者中显示出症状的缓解。然而,在SCD的临床试验中出现了两例急性髓系白血病(AML),尽管分析表明LV并非直接致病元凶,但患者体内存在的其他体细胞突变、慢性炎症的骨髓微环境以及重建应激可能共同增加了恶性克隆扩张的风险。
另一种策略是利用LV递送靶向BCL11A的短发夹RNA(shRNA),从而在红细胞系中特异性敲低BCL11A,重新激活内源性胎儿血红蛋白(HbF)的表达。初步临床试验显示部分患者HbF水平升高且症状减轻。此外,表达经过工程化改造以增强抗镰变活性的γ-珠蛋白(如GbGm LV)的策略,即使在较低载体拷贝数(VCN)下也能产生临床获益。
基因编辑技术的兴起为治疗带来了新维度。最成熟的策略是利用CRISPR-Cas9破坏BCL11A的红系增强子,从而解除对γ-珠蛋白基因的抑制,重新激活HbF的表达。基于此的exa-cel(Casgevy)疗法在针对Bthal和SCD的关键三期临床试验中表现出色,分别使绝大多数患者摆脱输血依赖或摆脱严重症状,并已获得美国FDA等监管机构的批准。类似地,在中国进行的一项临床试验通过编辑BCL11A增强子,也成功使儿科Bthal患者实现输血独立。其他编辑策略包括利用CRISPR-Cas12a靶向HBG1/2启动子区域,以及尝试通过同源定向修复(HDR)直接纠正SCD点突变,但后者因患者出现全血细胞减少症而暂停,提示药物产品制备工艺需进一步优化。
碱基编辑和引物编辑作为更精确的基因编辑工具,展现出潜在优势。碱基编辑器可以不引起DNA双链断裂(DSB)而实现单碱基转换,例如将SCD突变转换为良性的Hb Makassar变异,或在HBG启动子中模拟遗传性持续性胎儿血红蛋白(HPFH)突变。初步临床结果显示了高效靶向和HbF诱导能力。然而,其存在的旁观者编辑和潜在脱靶效应仍需在非临床研究中深入评估。
造血干细胞与骨髓微环境的生物学特征
尽管基因治疗取得了显著进展,但部分患者体内基因修饰HSC的植入水平较低,临床获益有限。这提示,疾病本身对HSC和其赖以生存的骨髓微环境的影响不容忽视。Bthal和SCD可被视为慢性应激状态下的范例,这种状态深刻改变了HSC的生物学特性和骨髓niche的功能。
在Bthal中,研究表明HSC数量减少,呈现出增殖活跃、压力增大和耗竭的迹象。其功能受损,移植后重建能力下降。分子层面显示,HSC中应激反应基因和DNA损伤标志物表达上调,而干性相关基因表达降低。慢性氧化应激和铁过载是导致HSC功能异常的关键因素,铁过载会引起HSC线粒体损伤,损害其代谢功能。此外,对接受基因治疗的Bthal患者进行的长时期谱系追踪分析发现,其造血过程优先向红系定向,即使在治疗后数年依然如此,这反映了疾病内在的造血压力。单细胞转录组分析进一步证实,Bthal患者的HSC/多能祖细胞(MPP)更活跃,干性降低,并向红系分化轨迹增强。
在SCD中,HSC同样表现出功能失调,包括增殖增加、DNA损伤和氧化应激。年轻SCD小鼠的HSC数量虽有轻度增加,但随年龄增长而减少,且其骨髓环境无法支持HSC的长期重建活性。慢性炎症和应激性红细胞生成影响了SCD患者CD34+ HSPCs的免疫表型和采集产量。表观遗传学研究还提示SCD患者存在加速生物老化现象,可能增加髓系恶性肿瘤的风险。在基因治疗临床试验中,对SCD患者来源的HSPCs进行分析发现,炎症信号通路(如IL-1、TNF-α和IFN通路)在最为原始的细胞组分中富集,这种炎症激活可能解释了基因修饰HSC植入不良的原因。
骨髓微环境,即HSC的“巢”,由基质细胞、可溶性因子和物理信号构成,对HSC的维持至关重要。在Bthal小鼠模型和患者中,骨髓微环境存在显著缺陷。表现为骨密度和骨沉积减少,骨系细胞(OLCs)和间充质基质细胞(MSCs)表达的关键niche分子(如骨桥蛋白、JAGGED1)减少,以及循环中甲状旁腺激素(PTH)水平下降。PTH-JAG1-Notch1信号轴的下调损害了OLCs/MSCs与HSC之间的通讯。此外,来自Bthal患者的MSCs会积累铁,表现出ROS水平升高、克隆形成能力和支持HSC的功能受损。另一个重要发现是,Bthal中由于高促红细胞生成素(EPO)刺激,成骨细胞和红系细胞产生的成纤维细胞生长因子23(FGF23)水平升高,后者会抑制PTH并负调控骨代谢,从而破坏HSC-niche的相互作用。在动物模型中,药理抑制FGF23信号可以改善骨病理并恢复HSC功能。
在SCD中,骨髓微环境同样受到破坏。SCD小鼠的MSCs频率降低,成骨和成脂分化能力受损,并表达下调多种关键的HSC支持因子(如Opn, Scf, Cxcl12)。骨髓血管网络也发生显著改变,表现为动脉迂曲、窦状血管碎片化,并伴有缺氧诱导因子1α(HIF-1α)水平升高和新生血管增加。慢性炎症细胞因子(如IL-6, IL-8, TNF-α)水平持续升高,可能通过上调RANKL促进破骨细胞活性,导致骨微结构异常和骨转换加速。随着年龄增长,SCD小鼠的MSCs会表现出促炎症基因表达增加,进一步削弱其支持HSC的能力。
对基因治疗的意义与未来方向
对HSC和骨髓微环境生物学的深入理解,为优化血红蛋白病基因治疗策略提供了新的视角。基因治疗的成功不仅取决于基因修饰的效率和安全性,也受到患者自身HSC质量和受体骨髓微环境状态的深刻影响。
在Bthal中,尽管无效红细胞生成和铁过载影响了HSC的自我更新和植入能力,但通过优化protocol和严格的安全性评估,基因治疗已显示出良好的风险获益比。在SCD中,针对HSC来源的氧化损伤和炎症特征进行干预,有望改善基因修饰细胞的植入并降低克隆性造血(CH)或恶性肿瘤的风险。
未来,开发联合治疗策略至关重要。这包括在基因治疗前后使用药物(如FGF23抑制剂、抗氧化剂)来改善骨髓微环境,使其更有利于基因校正HSC的定植和功能发挥。同时,优化HSC的体外培养条件以减轻应激损伤、保持干细胞特性,以及探索基于抗体药物偶联物(ADCs)或动员方案的清髓性预处理(conditioning)方案,以减少对骨髓微环境的损伤,都是重要的研究方向。
综上所述,血红蛋白病的基因治疗已经从概念验证走向临床现实。未来的治疗范式必将超越单纯的基因校正,转向结合针对缺陷的HSC及其微环境的综合性策略,旨在为患者实现持久、安全且彻底的治疗效果。
 生物通微信公众号
生物通微信公众号
 生物通新浪微博
生物通新浪微博
今日动态 |
人才市场 |
新技术专栏 |
中国科学人 |
云展台 |
BioHot |
云讲堂直播 |
会展中心 |
特价专栏 |
技术快讯 |
免费试用
版权所有 生物通
Copyright© eBiotrade.com, All Rights Reserved
联系信箱:
粤ICP备09063491号