FACT与RRM2双重抑制:靶向H2BG53D驱动胰腺癌的新策略
《Cell Reports》:Dual inhibition of FACT and RRM2 suppresses the progression of pancreatic ductal adenocarcinoma driven by the oncohistone H2BG53D
【字体:
大
中
小
】
时间:2025年11月24日
来源:Cell Reports 6.9
编辑推荐:
本研究针对胰腺导管腺癌(PDAC)中组蛋白突变H2BG53D的致癌机制展开探索。研究人员通过基因工程小鼠模型发现H2BG53D通过增强与FACT复合物的结合,改变转录谱促进PDAC发展。全基因组CRISPR-Cas9筛选鉴定RRM2为合成致死靶点,双重抑制FACT和RRM2显著延长小鼠生存期。该研究为H2BG53D突变PDAC提供了新的治疗策略。
胰腺导管腺癌(PDAC)是恶性程度最高的肿瘤之一,5年生存率不足10%。这种疾病的致命性源于其早期转移特性和对传统化疗的耐药性。近年来,表观遗传学异常在肿瘤发生发展中的作用日益受到关注,其中组蛋白突变作为新兴的致癌驱动因素备受瞩目。
在众多组蛋白突变中,H2BG53D在6.9%的胰腺癌病例中被发现,但其在体内的致癌机制和临床意义尚不明确。该突变位于H2B的球状结构域,不同于传统的组蛋白尾部突变,其通过改变核小体稳定性影响基因表达。然而,H2BG53D如何影响染色质功能并促进PDAC进展,以及是否存在针对该突变的特异性治疗策略,成为领域内亟待解决的关键科学问题。
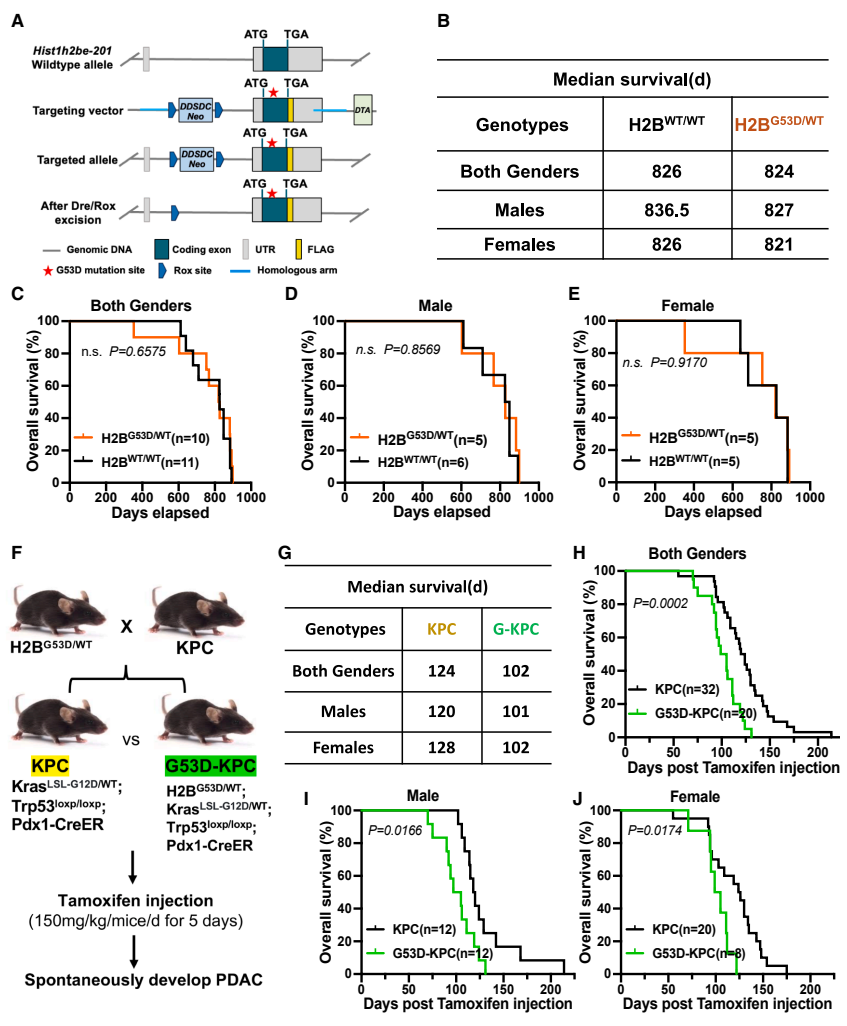
针对这些问题,陈居明教授团队在《Cell Reports》上发表了最新研究成果。研究人员首先构建了H2BG53D基因敲入小鼠模型,并将其与经典的KPC(KrasLSL-G12D/WT; Trp53LoxP/LoxP; Pdx1-CreER)胰腺癌模型杂交,系统评估了H2BG53D对PDAC发生发展的影响。
令人惊讶的是,单纯H2BG53D杂合突变并不影响小鼠生存,但在KPC背景下,该突变将小鼠中位生存期从124天显著缩短至102天。进一步分析发现,H2BG53D不仅促进原发肿瘤生长,还显著增加肝和膈肌转移发生率。RNA测序显示突变肿瘤中上皮-间质转化和缺氧通路激活,解释了其恶性表型的分子基础。
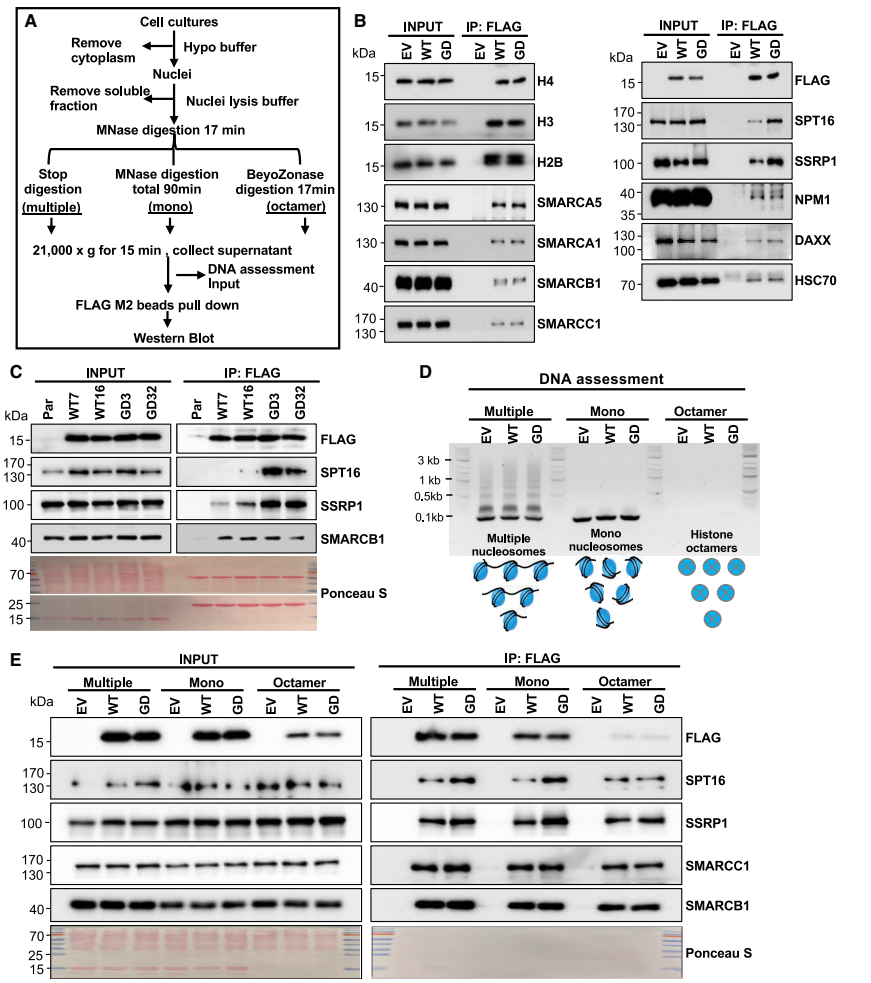
机制探索方面,研究人员通过免疫共沉淀技术发现H2BG53D特异性增强核小体与FACT复合物(包括SPT16和SSRP1亚基)的结合,而这种增强作用依赖于核小体DNA的存在。当研究人员敲低FACT复合物关键组分SPT16时,H2BG53D诱导的转录激活和细胞迁移能力被显著逆转,表明FACT在介导突变表型中起关键作用。
为寻找治疗靶点,团队进行了全基因组CRISPR-Cas9筛选,发现核糖核苷酸还原酶亚基M2(RRM2)是H2BG53D突变细胞的合成致死靶点。虽然RRM2抑制剂clofarabine在临床应用中存在毒性问题,但研究人员创新性地将其与FACT抑制剂curaxin联用,在体外实验中显示出对突变细胞的特异性杀伤作用。
在动物实验中,clofarabine(15 mg/kg)联合curaxin(7.5 mg/kg)的治疗方案将G53D-KPC小鼠的中位生存期从103天延长至135天,并显著降低肿瘤负荷。值得注意的是,这种联合治疗对野生型KPC小鼠效果有限,体现了其对H2BG53D突变肿瘤的特异性治疗潜力。
本研究主要采用基因工程小鼠模型、全基因组CRISPR-Cas9筛选、染色质免疫共沉淀、RNA测序、CUT&RUN测序等技术方法。研究样本包括患者来源的PDAC细胞系S2VP10及其H2BG53D敲入模型,以及KPC和G53D-KPC转基因小鼠模型。
通过系统比较KPC和G53D-KPC小鼠,研究人员发现H2BG53D突变显著增加肿瘤重量和体积,降低肿瘤分化程度。免疫组化显示突变肿瘤中Ki67和p-ERK信号增强,表明细胞增殖活性升高。这些结果证实H2BG53D在体内促进PDAC恶性进展。
尸检分析显示G53D-KPC小鼠肝转移发生率从16.7%上升至63.6%,膈肌转移从8.3%增至45.5%。转移灶转录组分析揭示MAPK/ERK通路激活和细胞周期相关基因下调,符合迁移-增殖悖论理论,解释了突变促进转移的分子机制。
H2BG53D elevates the interaction between nucleosome and the FACT complex
生化实验证明H2BG53D特异性增强与FACT复合物的结合,而不影响其他分子伴侣或染色质重塑因子。这种增强作用在多种核小体和单核小体条件下均存在,但在组蛋白八聚体水平消失,表明其DNA依赖性。
FACT is responsible for the H2BG53D-induced gene upregulation and oncogenic phenotypes
FACT缺失逆转H2BG53D引起的转录组改变,特别是那些富集突变核小体的基因。转录延伸实验显示FACT敲低显著降低ANXA3的转录速率。功能实验证实抑制FACT可逆转突变细胞的迁移增强表型。
Genome-wide CRISPR-Cas9 screen identified RRM2 as a promising therapeutic target
筛选鉴定出92个H2BG53D特异性依赖基因,其中RRM2的抑制剂clofarabine对突变细胞表现出选择性毒性。联合使用低剂量clofarabine和curaxin可在降低正常细胞毒性的同时有效抑制PDAC细胞生长。
Combinatory treatment suppresses H2BG53D-PDAC development
体内实验证明联合治疗显著延长小鼠生存期,减少原发和转移肿瘤负荷,且对野生型肿瘤效果有限,体现其靶向性治疗优势。
本研究首次在体内证实H2BG53D通过增强核小体-FACT相互作用促进PDAC发展和转移。机制上,该突变改变染色质结构,促进致癌基因转录激活。治疗方面,发现RRM2是合成致死靶点,双重抑制FACT和RRM2为H2BG53D突变PDAC提供新的治疗策略。该研究不仅阐明oncohistone的致病机制,还为精准治疗提供理论依据,具有重要临床转化价值。
 生物通微信公众号
生物通微信公众号
 生物通新浪微博
生物通新浪微博
今日动态 |
人才市场 |
新技术专栏 |
中国科学人 |
云展台 |
BioHot |
云讲堂直播 |
会展中心 |
特价专栏 |
技术快讯 |
免费试用
版权所有 生物通
Copyright© eBiotrade.com, All Rights Reserved
联系信箱:
粤ICP备09063491号