ЛљгкЖдБШбЇЯАгыУХПиОэЛ§ЩёОЭјТчЕФПЙОњыФдЄВтаТЗНЗЈЃКCG-AMPФЃаЭЕФЫЋФЃПщЬиеїШкКЯВпТдМАЦфзПдНадФм
ЁЖScientific ReportsЁЗЃКAntimicrobial peptide prediction based on contrastive learning and gated convolutional neural network
ЁОзжЬхЃК
Дѓ
жа
аЁ
ЁП
ЪБМфЃК2025Фъ11дТ25Ше
РДдДЃКScientific Reports 3.9
БрМЭЦМіЃК
ЁЁЁЁБОбаОПеыЖдПЙЩњЫиФЭвЉадШевцбЯжиЕФЙЋЙВЮРЩњЬєеНЃЌПЊЗЂСЫвЛжжЛљгкЩюЖШбЇЯАЕФПЙОњыФЃЈAMPЃЉЪЖБ№аТЙЄОпЁЊЁЊCG-AMPЁЃИУПђМмДДаТадЕиНсКЯСЫдЄбЕСЗгябдФЃаЭЃЈESM-2ЃЉЕФгявхЬиеїгыАќКЌНјЛЏаХЯЂЃЈBLOSUM62ЃЉКЭРэЛЏаджЪЃЈZ-scaleЃЉЕФЖрЮЌЪжЙЄЬиеїЃЌЭЈЙ§ЖдБШбЇЯАФЃПщКЭУХПиОэЛ§ЩёОЭјТчЃЈGCNNЃЉЫЋФЃПщМмЙЙНјааИпаЇЬиеїЬсШЁгыШкКЯЁЃдкAMPlifyКЭDAMPЖРСЂВтЪдМЏЩЯЃЌCG-AMPЕФзМШЗТЪЗжБ№ДяЕН0.9497КЭ0.9403ЃЌТэаоЫЙЯрЙиЯЕЪ§ЃЈMCCЃЉЗжБ№ЮЊ0.8994КЭ0.8812ЃЌЯджјгХгкЯжгажїСїЗНЗЈЃЌЮЊМгЫйаТаЭПЙОњыФЕФЗЂЯжЬсЙЉСЫПЩППЕФМЦЫуНтОіЗНАИЁЃ
ЁЁЁЁ
дкЙЋЙВЮРЩњСьгђЃЌПЙЩњЫиЕФЙ§ЖШЪЙгУЕМжТЯИОњФЭвЉадЮЪЬтШевцбЯОўЃЌвбГЩЮЊШЋЧђадЕФНЁПЕЭўаВЁЃИљОнУРЙњМВВЁПижЦгыдЄЗРжааФЃЈCDCЃЉЕФБЈИцЃЌНідкУРЙњЃЌУПФъОЭгадМ280ЭђР§гЩФЭвЉОњв§Ц№ЕФИаШОВЁР§ЃЌЕМжТГЌЙ§3.5ЭђШЫЫРЭіЁЃдкХЗжоЃЌУПФъдМга3.3ЭђШЫЫРгкФЭвЉОњИаШОЁЃвђДЫЃЌПЊЗЂФмЙЛЬцДњДЋЭГПЙЩњЫиЕФаТаЭПЙОњвЉЮяЦШдкУМНоЁЃПЙОњыФЃЈAntimicrobial Peptides, AMPsЃЉзїЮЊвЛжжОпгаЙуЦзПЙОњЛюадЕФаЁЗжзгыФЃЌФмЙЛжБНгАаЯђЮЂЩњЮяВЂЭљЭљЕМжТЯИАћСбНтЃЌЛђЭЈЙ§ЕїНкЫожїУтвпЯЕЭГРДдіЧПЖдЮЂЩњЮяЕФЗРгљФмСІЃЌЦфзїгУЫйТЪЭЈГЃБШДЋЭГПЙЩњЫиИќПьЁЃжЕЕУзЂвтЕФЪЧЃЌОЁЙмЯИОњдкЪ§АйЭђФъЕФНјЛЏЙ§ГЬжаГжајБЉТЖгкПЙОњыФЃЌЕЋВЂЮДВњЩњЯджјЕФФЭвЉадЃЌетЪЙЦфГЩЮЊМЋОпЧБСІЕФПЙЩњЫиЬцДњЦЗЁЃ
ШЛЖјЃЌЭЈЙ§ЪЊЪЕбщДѓЙцФЃЩИбЁаТаЭПЙОњыФВЛНіКФЪБЁЂАКЙѓЃЌЖјЧвРЭЖЏУмМЏаЭЁЃвђДЫЃЌдкЙ§ШЅМИЪЎФъжаЃЌЛљгкЛњЦїбЇЯАЕФМЦЫуЗНЗЈБЛЙуЗКгУгкМгЫйПЙОњыФЕФЪЖБ№ЃЌВЂШЁЕУСЫЯджјГЩЙћЁЃОЁЙмЯжгаЕФЛњЦїбЇЯАФЃаЭЃЈШчTriStackЁЂAMPlifyЕШЃЉвбеЙЯжГіСМКУЕФадФмЃЌЕЋЫќУЧШдДцдквЛаЉОжЯоадЃКЖрЪ§ЗНЗЈвРРЕгаЯоЕФЬиеїУшЪіЗћЃЌГЃГЃКіТдЙиМќЕФНјЛЏаХЯЂЁЂРэЛЏаджЪЛђЭЈЙ§дЄбЕСЗФЃаЭЬсШЁЕФЬиеїЃЛаэЖрЗНЗЈВЩгУМђЕЅЕФЬиеїШкКЯВпТдЛђЭГвЛЕФЭјТчМмЙЙНјааЬиеїЬсШЁЃЌЮДФмГфЗжПМТЧВЛЭЌЬиеїжЎМфЕФЛЅВЙадКЭЖрбљадЃЛДЫЭтЃЌГЄЖЬЦкМЧвфЭјТчЃЈLSTMЃЉЕШбЛЗЩёОЭјТчдкНЈФЃађСаЩЯЯТЮФЙиЯЕЪБВЮЪ§СПДѓЃЌПЩФмдкЖЬыФЬиеїЬсШЁЙ§ГЬжав§ШыШпграХЯЂЛђдіМгМЦЫуПЊЯњЃЌгАЯьФЃаЭЕФМЦЫуаЇТЪКЭЗКЛЏФмСІЁЃ
ЮЊСЫНтОіЩЯЪіЮЪЬтЃЌЗЂБэдкЁЖScientific ReportsЁЗЩЯЕФетЯюбаОПЬсГіСЫвЛжжаТгБЕФЫЋФЃПщЩёОЭјТчПђМмЁЊЁЊCG-AMPЃЌгУгкПЙОњыФЕФИпаЇЪЖБ№ЁЃИУПђМмЕФКЫаФДДаТдкгкгааЇећКЯСЫЖрдДЬиеїаХЯЂЃЌВЂВЩгУЖдБШбЇЯАКЭУХПиОэЛ§ЩёОЭјТчЃЈGated Convolutional Neural Network, GCNNЃЉСНжжгХЪЦЛЅВЙЕФФЃПщРДбЇЯАетаЉЬиеїЁЃ
баОПШЫдБЮЊПЊеЙДЫЯюбаОПЃЌжївЊгІгУСЫвдЯТМИЯюЙиМќММЪѕЗНЗЈЃКЪзЯШЃЌДгЙЋПЊШЈЭўЪ§ОнПтЃЈAPD3ЁЂDADPЁЂUniProtKB/Swiss-ProtЃЉЪеМЏВЂбЯИёЩИбЁСЫAMPlifyКЭDAMPСНИіЛљзМЪ§ОнМЏЃЌШЗБЃЪ§ОнЦНКтгыПЩППадЁЃЦфДЮЃЌВЩгУЖржжБрТыЗНЪНБэеїыФађСааХЯЂЃЌАќРЈРћгУESM-2дЄбЕСЗгябдФЃаЭЛёШЁ1280ЮЌЕФЩЯЯТЮФгявхЬиеїЃЌвдМАНсКЯBLOSUM62ЃЈНјЛЏаХЯЂЃЉЁЂZ-scaleЃЈРэЛЏаджЪЃЉКЭOne-HotБрТыЙЙНЈ45ЮЌЕФЖрЮЌЪжЙЄЬиеїЁЃЕкШ§ЃЌЩшМЦСЫЫЋФЃПщЬиеїбЇЯАМмЙЙЃЌЦфжаЖдБШбЇЯАФЃПщЪЙгУЖрВуИажЊЛњЃЈMLPЃЉБрТыЦїЃЌЭЈЙ§МрЖНЖдБШЫ№ЪЇКЏЪ§гХЛЏESM-2ЬиеїЕФБэеїПеМфЃЌдіЧПРрМфЧјЗжЖШЃЛGCNNФЃПщдђРћгУВЂааОэЛ§ЗжжЇЃЈКЫГпДч3,5,7ЃЉКЭУХПиЯпадЕЅдЊЃЈGLUЃЉЛњжЦИпаЇЬсШЁЖрЮЌЬиеїЕФађСаЩЯЯТЮФаХЯЂЃЌВЂв§ШыВаВюСЌНгЬсЩ§бЕСЗЮШЖЈадЁЃзюКѓЃЌЭЈЙ§ЯпадШкКЯВпТдећКЯЫЋФЃПщЪфГіЕФЬиеїЃЌВЂВЩгУШЋОжзюДѓГиЛЏНЕЮЌЃЌЪЙгУНЛВцьиЫ№ЪЇКЏЪ§НјааФЃаЭбЕСЗгыгХЛЏЁЃФЃаЭдкNVIDIA RTX 3090Ti GPUЩЯЛљгкPyTorchПђМмЪЕЯжЁЃ
CG-AMPФЃаЭВЩгУЫЋФЃПщМмЙЙЁЃЕквЛИіФЃПщзЈзЂгкбЇЯАдЄбЕСЗгябдФЃаЭЃЈESM-2ЃЉЬсЙЉЕФЬиеїБэЪОПеМфЃЌВЂЭЈЙ§ЖдБШбЇЯАНјаагХЛЏЁЃЕкЖўИіФЃПщдђВЩгУдіЧПЕФОэЛ§ЩёОЭјТчЃЈCNNЃЉЃЌОпЬхЮЊУХПиОэЛ§ЩёОЭјТчЃЈGCNNЃЉЃЌвдИќИпаЇЕиЬсШЁЪжЙЄЙЙНЈЕФЖрЮЌЬиеїЃЈАќРЈBLOSUM62ЁЂZ-scaleКЭOne-HotБрТыЃЉжаЕФаХЯЂЁЃетжжЩшМЦжМдкНсКЯСНжжЗНЗЈЕФгХЪЦЃЌгааЇећКЯЖрФЃЬЌЬиеїЃЌДгЖјЬсЩ§ПЙОњыФЪЖБ№ЕФзМШЗадКЭаЇТЪЁЃ
баОПВЩгУСЫЖржжБрТыЗНЗЈРДДгЖрНЧЖШЬсШЁађСааХЯЂЁЃESM-2ФЃаЭЮЊУПИіАБЛљЫсЩњГЩвЛИі1280ЮЌЕФЯђСПЃЌаЮГЩLЁС1280ЮЌЕФЬиеїОиеѓЃЈLЮЊађСаГЄЖШЃЉЁЃДЫЭтЃЌЛЙНЋOne-HotЃЈ20ЮЌЃЉЁЂBLOSUM62ЃЈ20ЮЌЃЉКЭZ-scaleЃЈ5ЮЌЃЉБрТыШкКЯЃЌЙЙНЈСЫвЛИіLЁС45ЮЌЕФЖрЮЌЬиеїОиеѓЁЃетжжЖрЪгНЧЕФЬиеїБэЪОЮЊФЃаЭЬсЙЉСЫЗсИЛЕФађСааХЯЂЁЃ
ЛљгкЖдБШбЇЯАЕФЬиеїБэЪОбЇЯА
ИУФЃПщВЩгУМрЖНЖдБШбЇЯАВпТдРДбЕСЗвЛИіMLPБрТыЦїЃЌвддіЧПдЄбЕСЗЬиеїЕФХаБ№ФмСІЁЃЦфКЫаФЪЧбЇЯАвЛИіЖШСППеМфЃЌЪЙЕУЭЌРрбљБОЃЈЭЌЮЊAMPЛђЭЌЮЊЗЧAMPЃЉЕФЧЖШыБЫДЫППНќЃЌЖјвьРрбљБОЕФЧЖШыЯрЛЅдЖРыЁЃЭЈЙ§ЖЈвхЖдБШЫ№ЪЇКЏЪ§ЃЌИУФЃПщЯдЪНЕибЇЯАРрБ№МЖБ№ЕФХаБ№адБэЪОЃЌЖјЗЧвРРЕгкЫцЛњЕФађСаШХЖЏЁЃ
ЛљгкУХПиОэЛ§ЩёОЭјТчЕФЬиеїЬсШЁ
GCNNећКЯСЫCNNЕФВЂааОэЛ§ВйзїКЭУХПиЯпадЕЅдЊЃЈGLUЃЉЕФУХПиЛњжЦЃЌЮЊађСаНЈФЃЬсЙЉСЫвЛИіИпаЇЖјЧПДѓЕФПђМмЁЃгыбаОПжаЪЙгУЕФLSTMЯрБШЃЌGCNNдкБЃГжЩѕжСЬсЩ§адФмЕФЭЌЪБЃЌОпгаИќИпЕФМЦЫуаЇТЪЁЃБОбаОПЩшМЦСЫвЛИіШ§ВуGCNNФЃПщЃЌУПВуАќКЌШ§ИіВЂааЕФ1DОэЛ§ЗжжЇЃЈКЫДѓаЁЗжБ№ЮЊ3ЁЂ5ЁЂ7ЃЉЃЌвдЪЕЯжЖрГпЖШИаЪмвАбЇЯАЁЃЭЈЙ§GLUЛњжЦКЭВаВюСЌНгЃЌИУФЃПщФмЙЛгааЇВЖЛёыФађСаЕФЩЯЯТЮФаХЯЂЃЌЭЌЪБЛКНтЬнЖШЯћЪЇЮЪЬтЃЌдіЧПбЕСЗЮШЖЈадЁЃ
ЮЊСЫгааЇШкКЯСНИіФЃПщбЇЯАЕНЕФЬиеїЃЌбаОПЪзЯШЖдУПИіФЃПщЬсШЁЕФађСаЬиеїЗжБ№гІгУШЋОжзюДѓГиЛЏвдНЕЕЭЬиеїЮЌЖШЃЌЫцКѓВЩгУЯпадШкКЯЛњжЦНсКЯГиЛЏКѓЕФЬиеїЁЃзюжеЕФдЄВтЗжЪ§ЭЈЙ§SigmoidКЏЪ§гГЩфЕН[0,1]ЧјМфЃЌВЂЪЙгУНЛВцьиЫ№ЪЇКЏЪ§РДЦРЙРФЃаЭЕФзМШЗадЁЃ
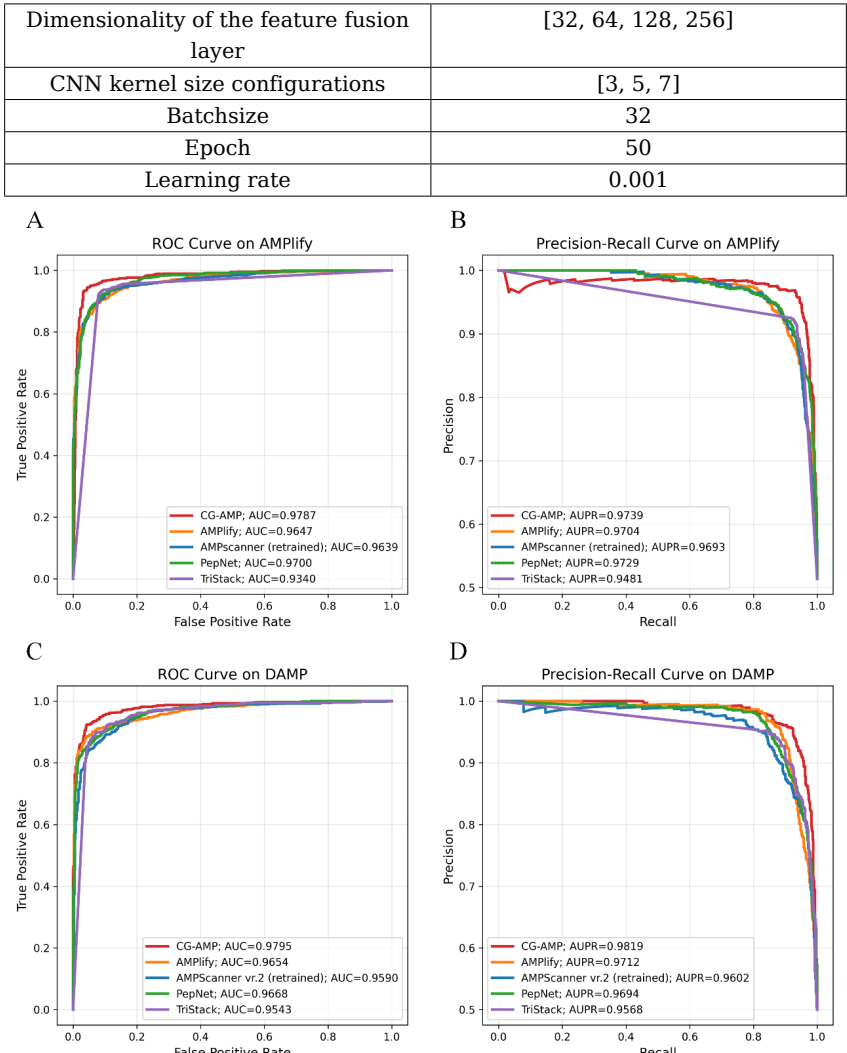
баОПШЫдБдкAMPlifyКЭDAMPСНИіЖРСЂВтЪдМЏЩЯЦРЙРСЫCG-AMPЕФадФмЃЌВЂгыЕБЧАзюЯШНјЕФФЃаЭЃЈАќРЈTriStackЁЂAMPlifyЁЂAMPscanner Vr2ЃЈжиаТбЕСЗЃЉКЭPepNetЃЉНјааСЫБШНЯЁЃЪЕбщНсЙћБэУїЃЌCG-AMPдкЖрИіЙиМќЦРЙРжИБъЩЯОљЯджјгХгкЦфЫћЖдБШФЃаЭЁЃОпЬхЖјбдЃЌдкAMPlifyВтЪдМЏЩЯЃЌCG-AMPЕФзМШЗТЪЃЈAccuracyЃЉДяЕН0.9497ЃЌF1ЗжЪ§ЃЈF1-scoreЃЉЮЊ0.9508ЃЌТэаоЫЙЯрЙиЯЕЪ§ЃЈMCCЃЉЮЊ0.8994ЁЃдкDAMPВтЪдМЏЩЯЃЌЦфзМШЗТЪЮЊ0.9403ЃЌF1ЗжЪ§ЮЊ0.9392ЃЌMCCЮЊ0.8812ЁЃДЫЭтЃЌНгЪеепВйзїЬиеїЧњЯпЃЈROCЧњЯпЃЉКЭОЋШЗТЪ-ейЛиТЪЧњЯпЃЈPRЧњЯпЃЉЕФПЩЪгЛЏЗжЮівВНјвЛВНжЄЪЕСЫCG-AMPдкПЙОњыФЪЖБ№ШЮЮёжаОпгаИќИпЕФЗжРрФмСІКЭЮШЖЈадЁЃ
ЮЊСЫЦРЙРCG-AMPИїИізщМўЕФЙБЯзЃЌбаОПНјааСЫвЛЯЕСаЯћШкЪЕбщЁЃЪЕбщЩшМЦСЫЫФИіБфЬхФЃаЭЃКCG-AMP-MЃЈНіБЃСєдЄбЕСЗЬиеїФЃПщЃЉЁЂCG-AMP-PЃЈНіБЃСєЖрЮЌЬиеїФЃПщЃЉЁЂCG-AMP-MCЃЈдкCG-AMP-MЛљДЁЩЯШЅГ§ЖдБШбЇЯАФЃПщЃЉКЭCG-AMP-PGЃЈдкCG-AMP-PЛљДЁЩЯШЅГ§GCNNФЃПщЃЉЁЃЪЕбщНсЙћБэУїЃЌЮоТлЪЧвЦГ§дЄбЕСЗЬиеїФЃПщЛЙЪЧЖрЮЌЬиеїФЃПщЃЌФЃаЭадФмЖМЛсГіЯжУїЯдЯТНЕЃЌетбщжЄСЫЫЋФЃПщЩшМЦЕФгааЇадЁЃЭЌЪБЃЌЖдБШбЇЯАФЃПщКЭGCNNФЃПщЕФвЦГ§вВЕМжТСЫадФмЕФЯджјНЕЕЭЃЌжЄУїСЫЫќУЧдкЬиеїЬсШЁжаЕФЙиМќзїгУЁЃДЫЭтЃЌМЦЫуаЇТЪЖдБШЯдЪОЃЌGCNNФЃПщЕФЭЦРэЪБМфКЭбЕСЗЪБМфОљдЖЕЭгкLSTMФЃПщЃЌЧвВЮЪ§СПИќЩйЃЌЭЙЯдСЫЦфИпаЇадЁЃ
баОПЛЙБШНЯСЫШ§жжВЛЭЌЕФЬиеїШкКЯЗНЗЈЃКЯпадШкКЯЁЂЦДНгШкКЯКЭЛљгкзЂвтСІЕФШкКЯЁЃЪЕбщНсЙћЗЂЯжЃЌЯпадШкКЯВпТдШЁЕУСЫзюМбадФмЁЃетБэУїЃЌОЙ§ЖдБШбЇЯАгХЛЏКѓЕФЬиеївбООпБИСЫКмЧПЕФХаБ№адКЭЛЅВЙадЃЌМђЕЅЕФЯпадМгШЈзувдЪЕЯжгааЇЕФЬиеїећКЯЃЌЖјИќИДдгЕФзЂвтСІЛњжЦПЩФмЛсвђв§ШыЙ§ЖрВЮЪ§ЖјЕМжТЙ§ФтКЯЁЃ
ЭЈЙ§t-SNEНЕЮЌММЪѕЖдCG-AMPФЃаЭбЇЯАЕНЕФЬиеїБэЪОНјааПЩЪгЛЏЗжЮіЁЃНсЙћЯдЪОЃЌгыДЋЭГЕФЖрЮЌЬиеїЯрБШЃЌдЄбЕСЗЬиеїдкРрБ№МфБэЯжГіИќУїЯдЕФЗжРыадЁЃОЙ§ЫЋФЃПщЬиеїШкКЯКѓЃЌФЃаЭбЇЯАЕНЕФЬиеїБэЪОЦфРрФкОлМЏЖШКЭРрМфЗжРыЖШЖМЕУЕНСЫЯджјдіЧПЃЌНјвЛВНжЄЪЕСЫCG-AMPФмЙЛбЇЯАЕНгааЇЧјЗжПЙОњыФгыЗЧПЙОњыФЕФЬиеїБэЪОЁЃ
ЮЊСЫбщжЄЫљЬсГіЕФCLЃЈЖдБШбЇЯАЃЉ+GCNNЫЋФЃПщМмЙЙЕФгааЇадЃЌбаОПНЋЦфгыСљжжЦфЫћЫЋФЃПщХфжУЃЈШчCL+BiLSTMЁЂCL+GCNЁЂCL+TransformerЕШЃЉНјааСЫБШНЯЁЃдкЫљгаХфжУжаЃЌCL+GCNNМмЙЙдкЖрИіЦРЙРжИБъЩЯ consistently БэЯжзюгХЃЌжЄУїСЫЦфдкПЙОњыФЪЖБ№ШЮЮёжаЕФзПдНгааЇадЁЃ
ЮЊСЫНјвЛВНЦРЙРФЃаЭЕФЗКЛЏФмСІЃЌбаОПНјааСЫСНЯюЖРСЂЕФЦРЙРЁЃЪзЯШЃЌЮЊСЫХХГ§ESM-2дЄбЕСЗЪ§ОнЃЈUniProtЃЉгыВтЪдМЏПЩФмДцдкЕФжиЕўЗчЯеЃЌбаОПШЫдБДгAMPlifyКЭDAMPВтЪдМЏжавЦГ§СЫгыUniRef50ЃЈESM-2ЕФбЕСЗЪ§ОнМЏЃЉжиЕўЕФађСаЃЌаЮГЩСЫвЛИіИЩОЛЕФЖРСЂВтЪдМЏЁЃдкИУВтЪдМЏЩЯЕФжиаТЦРЙРБэУїЃЌФЃаЭадФмБЃГжЮШЖЈЩѕжСТдгаЬсЩ§ЃЌжЄУїСЫЦфдкЭъШЋЮДМћЙ§ЕФађСаЩЯОпгаЮШНЁЕФЗКЛЏФмСІЁЃЦфДЮЃЌдквЛИіДгЮДдкбЕСЗЛђбщжЄжаЪЙгУЙ§ЕФЁЂРДздiAMP-bertЦРЙРЪ§ОнМЏЕФЖРСЂВтЪдМЏЩЯЃЌCG-AMPФЃаЭШЁЕУСЫ0.75ЕФзМШЗТЪЃЌЯджјИпгкЕкЖўУћTriStackФЃаЭЕФ0.62ЃЌНјвЛВНШЗШЯСЫЦфЧПДѓЕФХаБ№ФмСІКЭПчЪ§ОнМЏЕФЗКЛЏадФмЁЃ
БОбаОПЬсГіЕФCG-AMPПђМмЃЌЭЈЙ§ећКЯЖрдДађСаЬиеїЃЈдЄбЕСЗгявхЬиеїКЭАќКЌНјЛЏаХЯЂгыРэЛЏаджЪЕФЖрЮЌЪжЙЄЬиеїЃЉВЂВЩгУЖдБШбЇЯАгыУХПиОэЛ§ЩёОЭјТчЃЈGCNNЃЉЫЋФЃПщЬиеїЬсШЁВпТдЃЌГЩЙІЪЕЯжСЫЖдПЙОњыФЃЈAMPЃЉЕФИпОЋЖШЪЖБ№ЁЃИУФЃаЭЕФКЫаФгХЪЦдкгкЦфФмЙЛДгЖрИіЪгНЧГфЗжЭкОђыФађСаЕФЗсИЛаХЯЂЃЌВЂЭЈЙ§ИпаЇЕФШкКЯЛњжЦЪЕЯжЬиеїЛЅВЙЁЃЙуЗКЕФЪЕбщбщжЄБэУїЃЌCG-AMPдкЖрИіЛљзМВтЪдМЏЩЯОљЯджјгХгкЯжгажїСїЗНЗЈЃЌеЙЯжГігХвьЕФзМШЗадЁЂЮШЖЈадМАЧПДѓЕФЗКЛЏФмСІЁЃ
ИУбаОПЕФГЩЙІжївЊЙщвђгкСНИіЙиМќвђЫиЃКвЛЪЧШЋУцЖјЛЅВЙЕФЬиеїЙЄГЬЃЌКИЧСЫађСаЕФЩЯЯТЮФаХЯЂЁЂНјЛЏРњЪЗКЭЮяРэЛЏбЇЬиадЃЛЖўЪЧДДаТЕФЫЋФЃПщбЇЯАМмЙЙЃЌЪЙЕУгявхЬиеїКЭРэЛЏЬиеїФмЙЛЩюЖШШкКЯЃЌЦНКтСЫШЋОжгыОжВПвРРЕЙиЯЕНЈФЃЃЌДгЖјЩњГЩСЫИќОпаХЯЂСПЁЂИќЮШЖЈЧвИќЭЈгУЕФыФађСаБэЪОЁЃЯпадШкКЯВпТдБЛжЄУїЪЧЦНКтадФмгыИДдгадЕФгааЇбЁдёЁЃ
ОЁЙмФЃаЭадФмзПдНЃЌбаОПзїепвВжИГіСЫЮДРДПЩФмЕФгХЛЏЗНЯђЃЌР§ШчЭЈЙ§ИќДѓЙцФЃЕФбЕСЗНјвЛВНгХЛЏГЌВЮЪ§ЃЌЬНЫїМЏГЩЦфЫћдЄбЕСЗФЃаЭЃЈШчProtT5ЃЉЕФЬиеївдЛёШЁИќШЋУцЕФађСаБэЪОЃЌвдМАдкЮДРДИпОЋЖШыФНсЙЙдЄВтЙЄОпГЩЪьЪБЃЌГЂЪдећКЯыФЕФЖўМЖКЭШ§МЖНсЙЙаХЯЂЃЈШчІСТна§КЌСПЁЂЪшЫЎБэУцЛ§ЁЂНгДЅЭМЬиеїЕШЃЉРДдіЧПФЃаЭЖдПеМфЙЙЯѓКЭЙиМќЙІФмЮЛЕуЕФБэеїФмСІЁЃДЫЭтЃЌНЋCG-AMPПђМмЮЂЕїЛђгІгУгкЦфЫћЙІФмЕФыФЃЈШчПЙВЁЖОыФAVPsЁЂПЙАЉыФACPsЃЉЕФдЄВтШЮЮёЃЌвдЯЕЭГЦРЙРЦфПчШЮЮёЪЪгІадКЭПЩЧЈвЦадЃЌвВЪЧЮДРДЕФживЊбаОПЗНЯђЁЃ
злЩЯЫљЪіЃЌCG-AMPзїЮЊвЛИізЈзЂгкПЙОњыФЪЖБ№ЕФИпадФмМЦЫуЙЄОпЃЌЦфСщЛюЕФМмЙЙЪЙЦфОпгаСМКУЕФРЉеЙЧБСІЃЌВЛНіЮЊМгЫйаТаЭПЙОњыФЕФЗЂЯжЬсЙЉСЫЧПгаСІЕФММЪѕжЇГжЃЌвВЮЊНтОіађСаЧ§ЖЏЕФЩњЮяЗжРрШЮЮёЬсЙЉСЫПЩНшМјЕФЗЖЪНЁЃЫцзХШЫЙЄжЧФмММЪѕЕФГжајЗЂеЙЃЌетРрШкКЯЖрдДаХЯЂКЭЯШНјЩюЖШбЇЯАМмЙЙЕФФЃаЭЃЌгаЭћдкЩњЮявНбЇбаОПСьгђЗЂЛгдНРДдНживЊЕФзїгУЁЃ
 ЩњЮяЭЈЮЂаХЙЋжкКХ
ЩњЮяЭЈЮЂаХЙЋжкКХ
 ЩњЮяЭЈаТРЫЮЂВЉ
ЩњЮяЭЈаТРЫЮЂВЉ
- ЫбЫї
- ЙњМЪ
- ЙњФк
- ШЫЮя
- ВњвЕ
- ШШЕу
- ПЦЦе
НёШеЖЏЬЌ |
ШЫВХЪаГЁ |
аТММЪѕзЈРИ |
жаЙњПЦбЇШЫ |
дЦеЙЬЈ |
BioHot |
дЦНВЬУжБВЅ |
ЛсеЙжааФ |
ЬиМлзЈРИ |
ММЪѕПьбЖ |
УтЗбЪдгУ
АцШЈЫљга ЩњЮяЭЈ
Copyright© eBiotrade.com, All Rights Reserved
СЊЯЕаХЯфЃК
дСICPБИ09063491КХ