
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
基于染色体尺度长度变异(CSLV)的乳腺癌风险预测模型优化研究
【字体: 大 中 小 】 时间:2025年06月12日 来源:Human Genomics 3.8
编辑推荐:
本研究针对现有基于SNP(单核苷酸多态性)的乳腺癌多基因风险评分(PRS)预测精度不足(AUC仅0.60)且存在种族差异的问题,开发了基于染色体尺度长度变异(CSLV)的新型风险预测方法。通过分析NIH"All of Us"项目中49,051名女性的基因组数据(含4,533例乳腺癌患者),研究人员利用88个CSLV参数构建机器学习模型,将预测AUC提升至0.70(95% CI 0.67-0.73),高危组患病风险达低危组的9倍。该成果发表于《Human Genomics》,为跨种族乳腺癌早期筛查提供了更精准的工具。
乳腺癌是全球女性健康的主要威胁,每年仅美国就有28.7万新发病例和4.3万死亡病例。尽管早期诊断可显著提高生存率,但现有遗传检测手段存在明显局限:BRCA1/2突变仅覆盖5-10%的病例,而基于单核苷酸多态性(SNP)的多基因风险评分(PRS)预测效能有限(AUC约0.60),且在非欧裔人群中表现更差。更棘手的是,传统PRS需要百万级SNP数据和超大样本量,难以在少数族裔中推广应用。
针对这些挑战,Yasaman Fatapour和James P.Brody团队创新性地提出染色体尺度长度变异(CSLV)概念。这种将基因组表征为88个参数(每条常染色体4个区段的平均log R比值)的方法,巧妙规避了SNP分析的高维度难题。研究利用NIH"All of Us"项目49,051名女性数据(含440名非裔患者),通过H2O AutoML平台训练梯度提升机(GBM)等模型,取得三大突破性发现:
关键方法
研究结果
模型性能提升
将基因组表征从22个(全染色体均值)增至88个参数后,AUC从0.60显著提升至0.70。如图2所示,精细化的染色体分段能捕捉更丰富的结构变异信息。

临床实用价值
如表2所示,模型将测试集女性按风险分为五等份时,最高风险组(前20%)患癌几率是最低组的9倍(OR=3.47 vs 0.39)。这种分级能力可为精准筛查提供依据。
跨种族适用性
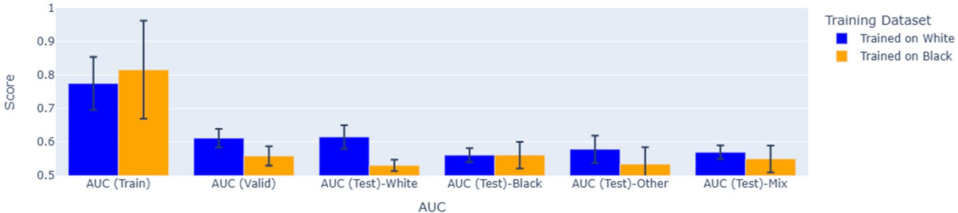
如图5所示,白人训练模型在白人测试集表现最佳(AUC 0.71±0.02),但在非裔测试集与非裔训练模型无显著差异(AUC 0.57±0.03)。SHAP分析(图4)显示风险预测依赖多染色体协同作用,而非单一热点区域。
结论与意义
该研究开创性地证明CSLV可作为乳腺癌风险预测的高效生物标志物,其优势体现在三方面:
这项发表于《Human Genomics》的研究,为破解"SNP数据依赖困境"提供了范式转换方案。未来通过整合SNP与CSLV数据、扩大少数族裔样本量,有望进一步优化预测效能,推动精准预防医学发展。
生物通微信公众号
知名企业招聘

