
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
Charm:基于Hi-C数据模拟染色体结构变异的创新计算框架及其在基因组研究中的应用
【字体: 大 中 小 】 时间:2025年06月20日 来源:NAR Genomics and Bioinformatics 4.0
编辑推荐:
为解决染色体结构变异(SV)检测中缺乏高质量模拟数据的难题,俄罗斯科学院西伯利亚分院细胞与遗传学研究所团队开发了Charm计算框架,该工具通过整合Hi-C数据的距离依赖性和位点特异性偏好,实现了染色体易位、倒位和拷贝数变异(CNV)的高保真模拟。研究证实Charm生成的仿真数据与真实实验数据相关性达0.85-0.95,并成功应用于EagleC、HiSV等SV检测工具的基准测试,为基因组三维结构研究提供了重要方法学突破。
基因组结构变异(SV)是驱动物种进化和人类疾病的关键因素,但传统检测技术难以精确识别平衡性重排的断点位置。近年来,染色质构象捕获技术(3C)及其衍生方法如Hi-C的出现为SV检测带来了新机遇,然而该领域面临一个根本性挑战――缺乏包含已知SV特征的标准化数据集,这严重制约了计算方法的开发和验证。
针对这一瓶颈,俄罗斯科学院西伯利亚分院细胞与遗传学研究所、新西伯利亚国立大学等机构的研究团队在《NAR Genomics and Bioinformatics》发表了创新性解决方案。研究人员开发了Charm(Chromosome rearrangement modeler)计算框架,该工具通过四步建模流程:参考数据统计计算、基因组坐标转换、接触计数预测和随机化处理,实现了染色体重排的高精度模拟。关键技术包括基于参考Hi-C数据建立距离依赖模型(计算OEAB值)、采用"平方根"和"求和"模型处理稀疏数据,以及通过二项分布模拟实验噪声。
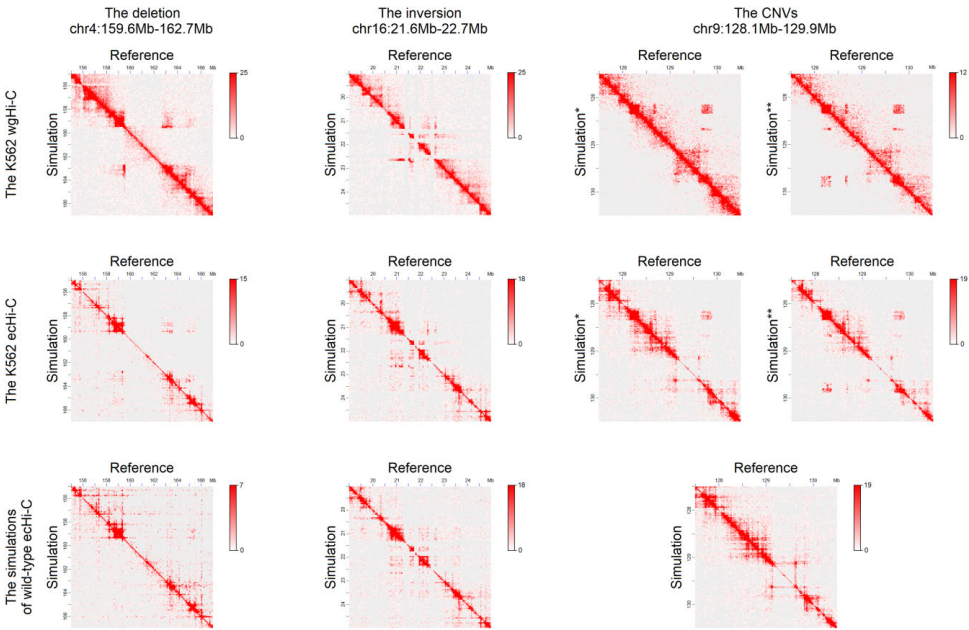
研究团队首先从参考Hi-C数据提取三大核心参数:基因组距离依赖的接触频率(Av(bi,bj))、位点特异性偏好(OE(bi,bj))和覆盖度偏差(covmult)。通过独创的坐标转换算法(R(bi,b?k)=mi(bi,b?k)×mi(b?k,bi)),Charm能准确映射重排前后基因组区域的对应关系。针对不同Hi-C技术特点,工具提供三种接触计数预测模型,其中外显子捕获Hi-C(ecHi-C)采用"求和"模型,全基因组Hi-C(wgHi-C)默认使用"平方根"模型。
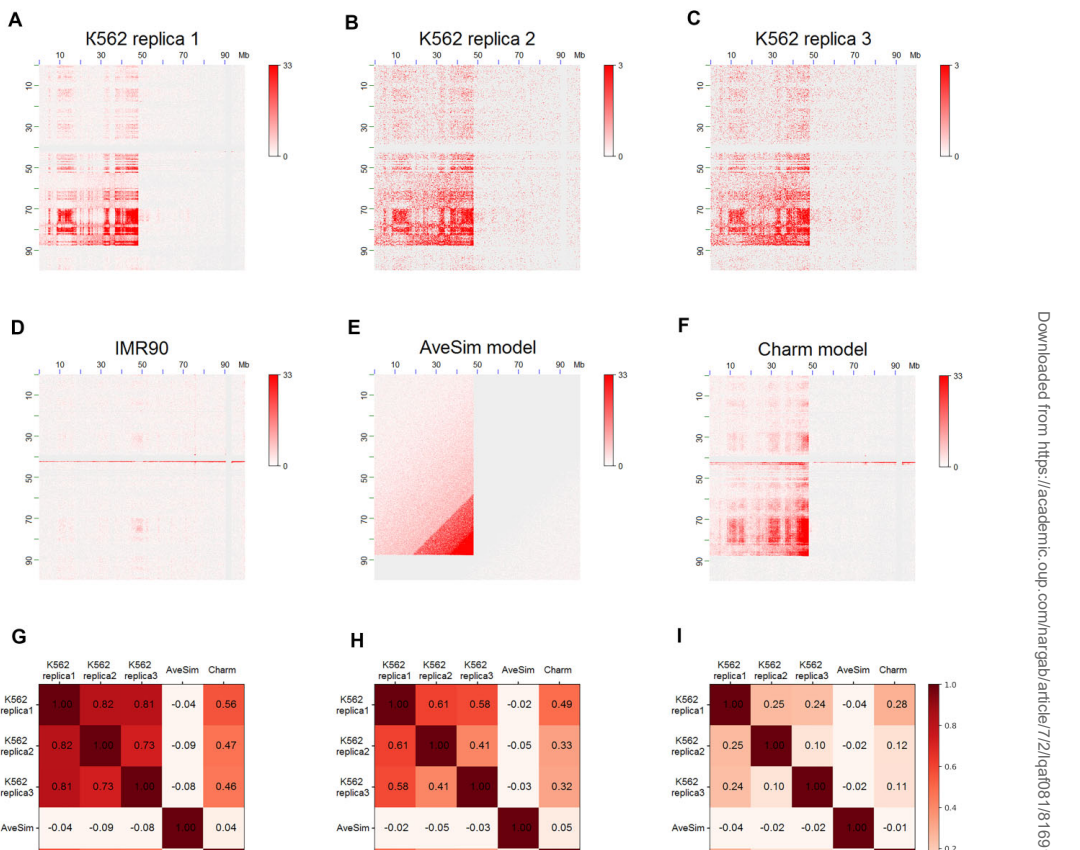
通过模拟K562细胞系已知SV(如chr4缺失、chr16倒位),Charm生成的数据与实验观测值Pearson相关系数达0.505-0.624,显著优于仅考虑距离效应的AveSim工具。研究创建了包含4400种SV的公共数据集(1760例易位、880例倒位、1760例CNV),尺寸覆盖100kb-20Mb范围。利用该数据集对EagleC、HiSV等工具进行基准测试发现:在易位检测中,HiSV召回率约50%且假阳性较低,而EagleC虽灵敏度相当但假阳性较高;对于倒位和CNV,所有工具性能均下降,wgHi-C数据上的表现普遍优于ecHi-C。
这项研究突破了染色体重排模拟的技术瓶颈,其创新性体现在三方面:首先,Charm是首个同时整合距离依赖性和位点特异性的Hi-C模拟器,能保留染色质区室化(A/B compartments)等三维架构特征;其次,工具采用模块化设计,允许用户自定义随机化函数(二项/正态/超几何分布)和分辨率(5-50kb);最重要的是,研究提供的标准化数据集为SV检测算法开发建立了金标准。这些成果将加速基于3C技术的基因组结构研究,特别是在癌症基因组学和进化遗传学领域具有重要应用价值。研究也指出当前工具的局限性――对短片段CNV(<500kb)和复杂重排的检测能力仍需提升,这为未来方法学改进指明了方向。
 生物通微信公众号
生物通微信公众号
知名企业招聘

