
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
基于全细胞-核质分离RNA测序的转录本亚细胞定位精准量化方法研究
【字体: 大 中 小 】 时间:2025年06月22日 来源:NAR Genomics and Bioinformatics 4.0
编辑推荐:
本研究通过开发新型贝叶斯回归模型,结合全细胞、核质分离RNA测序(RNAseq)数据,首次实现真核细胞转录本核质分布比例的精准计算。团队建立的定位指数(LI)模型突破传统FPKM比较法的局限,在模拟数据和ENCODE单细胞数据验证中显示优越性能,揭示保留内含子转录本核滞留机制及GC含量等序列特征与定位的关联,为基因表达调控研究提供新范式。
在真核细胞中,RNA分子在细胞核与细胞质间的分布调控是基因表达的关键环节。核滞留可抑制蛋白质翻译,而胞质定位则促进蛋白合成。传统原位杂交技术通量低,计算预测模型依赖序列特征易忽略环境特异性调控。更棘手的是,常规核质分离RNA测序数据因测量基准不同(核质RNA总量未知),导致FPKM值无法直接比较――这正是制约亚细胞RNA定位研究的核心瓶颈。
巴塞罗那科学技术研究所的Vasilis F. Ntasis和Roderic Guigó团队在《NAR Genomics and Bioinformatics》发表创新性研究,通过数学推导证明:当同时具备全细胞、核质RNA测序数据时,可准确计算胞质RNA体积占比β,进而建立定位指数(LI)量化单个转录本的核质分布。研究利用Flux模拟器生成β值已知的基准数据集,结合ENCODE项目的11种细胞系数据,发现70-90%多聚腺苷酸化RNA位于胞质,并首次系统揭示不同转录本类型(如保留内含子转录本与蛋白编码转录本)的定位偏好性及其序列特征关联。
关键技术包括:1) 使用Flux模拟器生成含预设β值(0.5-0.8)的模拟RNAseq数据;2) 整合ENCODE项目中9种细胞系和2种原代细胞的匹配全细胞-核质RNAseq数据;3) 开发基于Stan的贝叶斯线性回归模型(误差项采用t分布),通过FPKMw=(1-β)FPKMn+βFPKMc方程估算β值;4) 应用IRFinder和IPSA-NF分析内含子保留(IR-ratio)与外显子跳跃(PSI)事件。
理论模型构建
通过数学推导建立核心方程:FPKMw(i)=(1-β)FPKMn(i)+βFPKMc(i),证明β反映胞质RNA体积占比。如图1所示,当β偏离0.5时,传统"naive LI"(FPKMc/(FPKMn+FPKMc))会产生显著偏差,而新模型能准确还原模拟设定的分布比例。
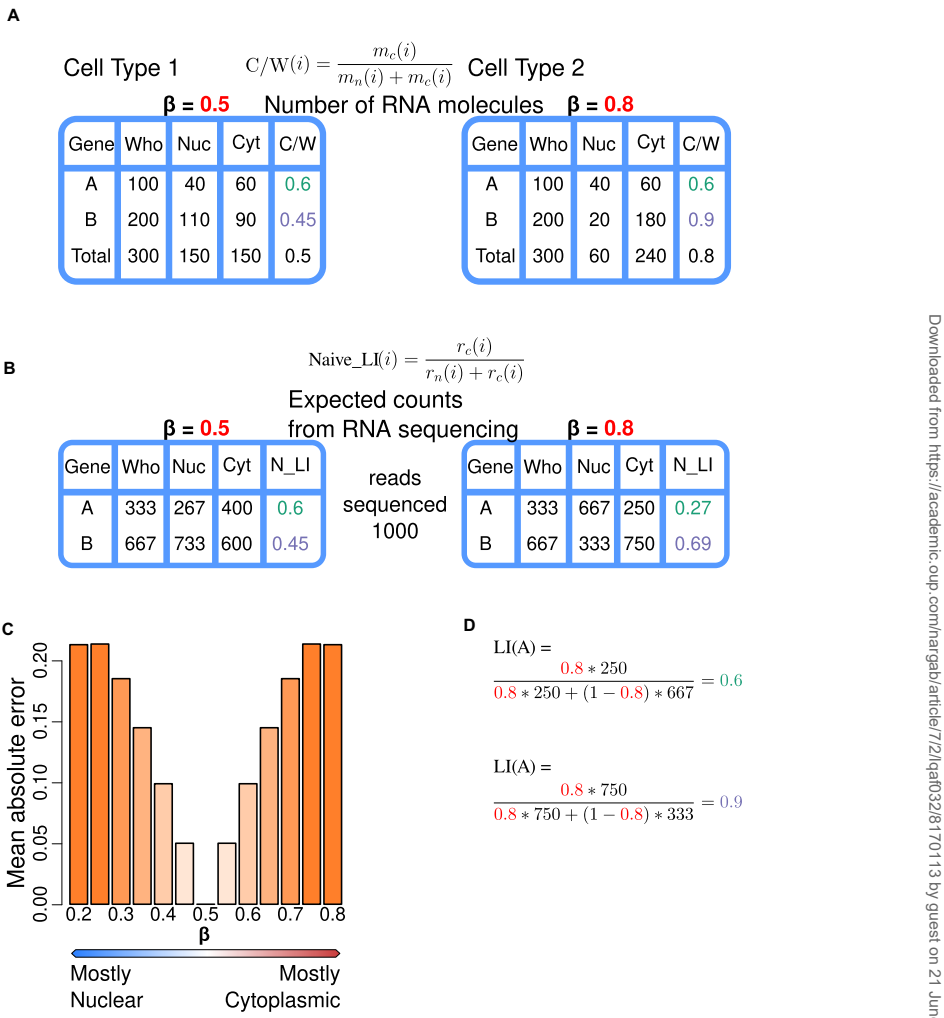
方法验证
在β值预设为0.5-0.8的模拟数据中,贝叶斯回归估算的β误差趋近于零(图2A),而传统方法误差随β偏离0.5显著增大(图2B)。单细胞数据测试显示,本方法估算β=0.82,更接近实验测量值0.84。
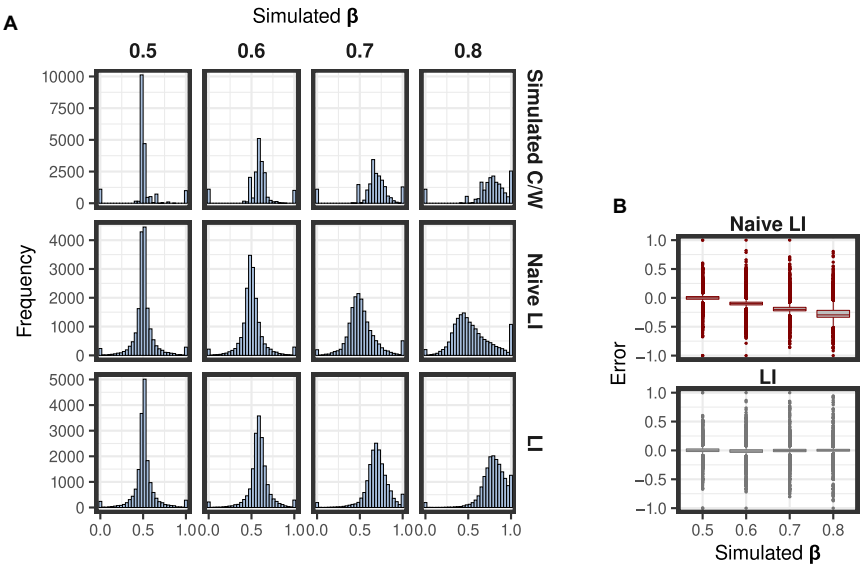
生物学发现

序列特征关联:核定位转录本具有更高GC含量(P<10-15)和更短内含子(图4C),其核特异性外显子的低PSI值(<0.5)富集度达2.1倍(图4D),提示剪接效率与定位调控的耦合。
跨细胞系保守性:72%的泛表达转录本(10细胞系)保持胞质定位一致性,显著高于条件特异性转录本(P<10-6)。
这项研究建立的定量框架解决了核质RNA测序数据不可比的长期难题。发现定位调控具有转录本特异性(同一基因不同异构体可分布在不同区室),且受剪接效率与序列特征共同影响。技术层面,贝叶斯模型对测序深度降低的稳健性(10%数据量时误差仅0.01)使其适用于广泛数据集。生物学意义上,揭示核滞留可能是通过内含子保留实现调控的新机制,为理解神经肌肉疾病等RNA定位异常相关疾病的病理提供新视角。
生物通微信公众号
知名企业招聘

