
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
连接酶诱导的DNA自组装新策略:从瞬态配对到稳定纳米结构的构建
【字体: 大 中 小 】 时间:2025年07月05日 来源:Nucleic Acids Research 16.7
编辑推荐:
本文针对传统DNA自组装中短链粘性末端(5-nt以下)结合不稳定的问题,提出了一种连接酶诱导的主动组装策略。研究通过T4 DNA连接酶将瞬态碱基配对转化为永久共价连接,成功构建了离散型四臂连接体(4-arm junction)二维晶格(2×2至6×6)、一维延伸结构及多级组装体(如雪片状DNA折纸单元)。该方法突破了常规杂交组装的温度限制(可在4°C实现),显著提升了短粘性末端(3-nt至5-nt)的组装效率与结构稳定性,为复杂DNA纳米结构的精准构建提供了新范式。
DNA纳米技术凭借沃森-克里克碱基配对规则,已实现从简单双链到复杂折纸结构的精准构建。然而,传统组装依赖长链粘性末端(通常>8 nt)的稳定杂交,短链粘性末端(≤5 nt)因结合力弱难以维持结构稳定性,严重限制了纳米结构的微型化与动态调控能力。现有技术中,DNA连接酶(ligase)多用于组装后稳定化处理,其主动参与组装过程的潜力尚未充分开发。如何利用酶促反应将瞬态结合转化为永久连接,成为突破短链组装瓶颈的关键挑战。
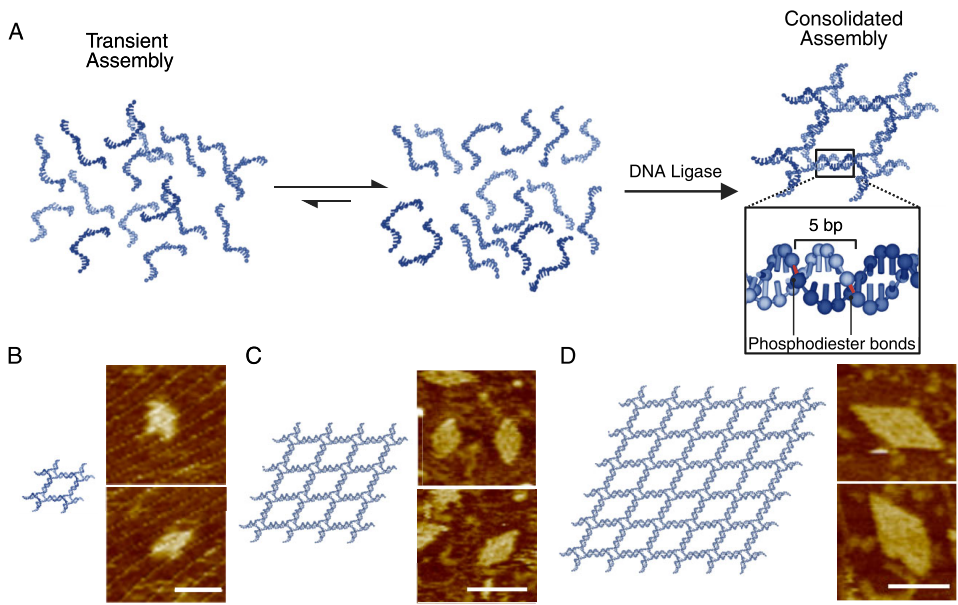
清华大学与华大研究院合作团队提出"连接酶诱导自组装"(ligation-induced assembly)新方法(图1)。核心策略是设计含3-nt至5-nt粘性末端的DNA元件(如四臂连接体、雪片状折纸单元),通过T4多核苷酸激酶(PNK)进行5'端磷酸化修饰,随后在T4 DNA连接酶催化下实现共价连接。关键技术包括:

设计含5-nt粘性末端的四臂连接体单元(图2A)。无连接酶时,2×2至6×6晶格在20-35°C均无法组装(凝胶电泳无目标条带);添加T4 DNA连接酶后,AFM清晰显示成功构建的晶格结构(图2B-D)。其中6×6晶格边长约100 nm,验证了方法的可扩展性。
基于5个四臂连接体单元构建重复单元(图3A),单元内以10-11 bp稳定配对,单元间通过5对4-nt粘性末端连接。连接酶诱导后形成宽度一致、长度达1 μm的带状结构(图3B),显著优于无酶对照组(无目标产物)。

预组装的5×5晶格单元通过边界4-nt粘性末端二次连接(图4A),成功构建I型三聚体、L型三聚体、T型四聚体及X型五聚体(图4B-E)。凝胶电泳显示连接酶组的产率(>60%)显著高于传统12-nt粘性末端组装(<30%),AFM证实结构完整性。

雪片状DNA折纸(snowflake-shaped origami)单元(图4F)通过3-nt粘性末端连接。在4°C低温下,T4 DNA连接酶成功催化形成三角形三聚体(图4G)与菱形四聚体(图4H),TEM显示精确的几何排布,突破常规组装温度限制。

本研究首创的连接酶诱导自组装策略,通过将瞬态碱基配对(3-nt至5-nt)转化为共价连接,实现了三类突破:
生物通微信公众号
知名企业招聘

