
-

ЩњЮяЭЈЙйЮЂ
ХуФузЅзЁЩњУќПЦММ
ЬјЖЏЕФТіВЋ
ЩњЮяЭЈЙйЮЂ
ХуФузЅзЁЩњУќПЦММ
ЬјЖЏЕФТіВЋ
ЖЬЖСГЄКъЛљвђзщЗжРрЙЄОпдкЗЯЫЎДІРэЮЂЩњЮяШКТфжаЕФадФмМЋЯоВтЪд
ЁОзжЬхЃК Дѓ жа аЁ ЁП ЪБМфЃК2025Фъ07дТ06Ше РДдДЃКScientific Reports 3.8
БрМЭЦМіЃК
ЁЁЁЁБОбаОПеыЖдЗЯЫЎДІРэЯЕЭГжаЮЂЩњЮяШКТфЗжРрЕФзМШЗадФбЬтЃЌЭЈЙ§ЙЙНЈФЃФтШКТфЃЈmock communityЃЉЃЌЯЕЭГЦРЙРСЫKaijuЁЂKraken2ЁЂRiboFrameКЭkMetaShotЫФжжЗжРрЙЄОпдкЖЬЖСГЄЃЈ150 bpЃЉЪ§ОнЯТЕФБэЯжЁЃбаОПЗЂЯжKaijuдкЪє/жжЫЎЦНЗжРрОЋЖШзюИпЃЌЕЋЫљгаЙЄОпОљДцдкЯджјЮѓЗжРрЗчЯеЃЌгШЦфецКЫгыЯИОњађСаЕФНЛВцДэЮѓПЩФмЮѓЕМЙиМќЮЂЩњЮяЙІФмбаОПЁЃИУГЩЙћЮЊЗЯЫЎДІРэЮЂЩњЮязщбаОПЬсЙЉСЫЗНЗЈбЇЛљзМЃЌЗЂБэгкЁЖScientific ReportsЁЗЁЃ
ЙЄвЕЛЏНјГЬдкЭЦЖЏЩчЛсЗЂеЙЕФЭЌЪБЃЌвВДјРДСЫбЯОўЕФЫЎЮлШОЬєеНЁЃЛюадЮлФрЃЈASЃЉКЭКУбѕПХСЃЮлФрЃЈAGSЃЉзїЮЊжїСїЩњЮяЗЯЫЎДІРэММЪѕЃЌЦфКЫаФдкгкИДдгЮЂЩњЮяШКТфЕФаЭЌзїгУЁЃШЛЖјЃЌетаЉЯЕЭГжаЮЂЩњЮязщГЩгыЙІФмЕФОЋШЗНтЮіГЄЦкЪмЯогкКъЛљвђзщЗжРрЙЄОпЕФзМШЗадЦПОБЁЊЁЊЯжгаЙЄОпЖрдкШЫРрЮЂЩњЮязщжабщжЄЃЌЖдИЛКЌЯЁгаЮяжжКЭЛЗОГЬивьадЮЂЩњЮяЕФЗЯЫЎЬхЯЕЪЪгУадДцвЩЁЃ
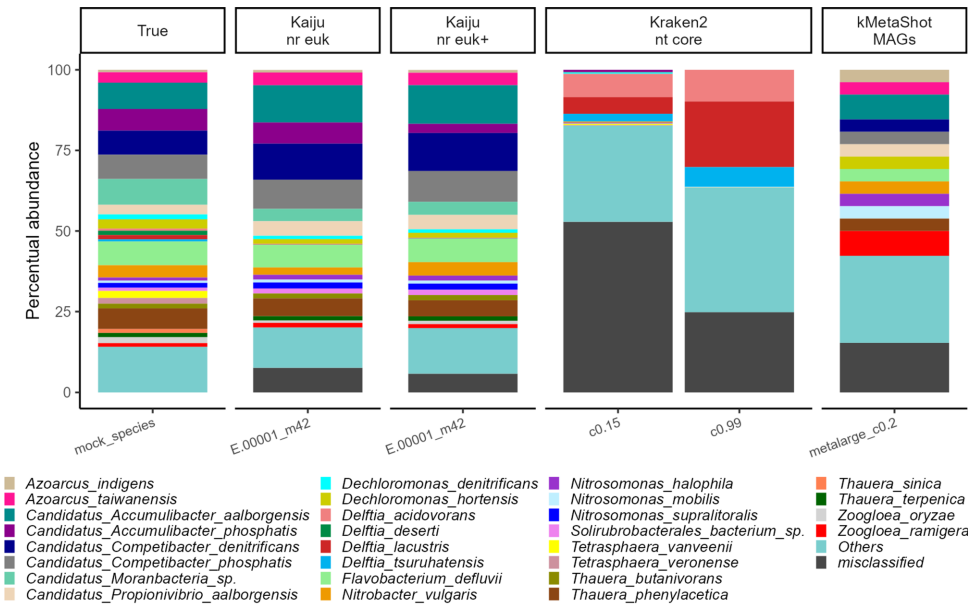
З№ТоТзШјДѓбЇЕФбаОПЭХЖгдкЁЖScientific ReportsЁЗЗЂБэСЫвЛЯюПЊДДадбаОПЃЌЭЈЙ§ОЋаФЩшМЦЕФФЃФтШКТфЃЈАќКЌ16жжЯИОњЁЂ3жжецКЫЩњЮяКЭT4ЪЩОњЬхЃЉЃЌЪзДЮЯЕЭГЦРЙРСЫЖЬЖСГЄКъЛљвђзщЗжРрЙЄОпдкЗЯЫЎЮЂЩњЮязщжаЕФадФмБпНчЁЃбаОПЗЂЯжЃКЕААзжЪЫЎЦНЗжРрЙЄОпKaijuЃЈnr euk+Ъ§ОнПтЃЉдкЪє/жжЗжРрзМШЗТЪЃЈ75%ЃЉКЭШКТфНсЙЙЛЙдЖШЩЯБэЯжзюгХЃЌЕЋЦфШдДцдкНЋШЫРрађСаЮѓХаЮЊХБдГцЃЈPlasmodium ovaleЃЉЕШбЯжиДэЮѓЃЛЖјЛљгкk-merЕФKraken2ЃЈnt coreЪ§ОнПтЃЉЫфФмМьГіШЋВПФПБъОњЪєЃЌЕЋМйбєадТЪИпДя25%ЁЃСюШЫвтЭтЕФЪЧЃЌзЈЮЊКъЛљвђзщзщзАЛљвђзщЃЈMAGsЃЉЩшМЦЕФkMetaShotдкMAGsВуУцЪЕЯжСуЮѓХаЃЌЕЋСщУєЖШНі30%ЃЌЭЙЯдзщзАжЪСПЖдЗжРраЇЙћЕФОіЖЈадгАЯьЁЃ
баОПВЩгУЖрЮЌЖШММЪѕТЗЯпЃК1ЃЉЪЙгУInSilicoSeqФЃФтIllumina NovaSeq 150 bpЫЋЖЫЖСГЄЪ§ОнЃЛ2ЃЉЭЈЙ§BBDukНјаажЪСППижЦЃЛ3ЃЉВЩгУMEGAHITШ§жжФЃЪНЃЈdefault/metalarge/customЃЉзщзАcontigsЃЛ4ЃЉРћгУMetaBat2ЙЙНЈMAGsЃЛ5ЃЉНЛВцБШНЯЫФжжЗжРрЙЄОпЃЈKaijuЁЂKraken2ЁЂRiboFrameЁЂkMetaShotЃЉдкreads/contigs/MAGsШ§ИіВуУцЕФБэЯжЃЛ6ЃЉв§ШыEukDetectКЭBowtie2ЦРЙРецКЫађСаЪЖБ№аЇФмЁЃ
Mock processing stats
жЪСППижЦКѓ92.6%ЕФЖСГЄНјШыЗжЮіЃЌИїЙЄОпЗжРраЇТЪВювьЯджјЃКKaijuЗжРрТЪ76-94%ЃЈШЁОігкзюаЁИВИЧуажЕmЃЉЃЌKraken2ЃЈnt coreЃЉдкжУаХуажЕ0.05ЪБЗжРр51%ЖСГЄЃЌЖјSILVAЪ§ОнПтЯТВЛзу2%ЁЃRiboFrameФкДцеМгУзюЕЭЃЈ20 GBЃЉЃЌЕЋНіФмЗжЮі16S rRNAЦЌЖЮЁЃ
Comparison at genus-level classification

Comparison at species-level classifications
Classification performances of phage T4 and lower metazoan
ецКЫЬивьадЙЄОпEukDetectСщУєЖШВЛзуЃЈНіМьГі23ЬѕDiploscapterЖСГЄЃЉЃЌЖјKaijuЃЈnr euk+ЃЉЫфФмЪЖБ№ЯпГцађСаЃЌЕЋДэЮѓЕиНЋЯИОњЖСГЄЙщРрЮЊSteinernemaЃЈРЅГцВЁдЯпГцЃЉЁЃT4ЪЩОњЬхЗжРржаЃЌKraken2ЃЈжУаХЖШ0.05ЃЉМьГіТЪ67%ЃЌЯджјгХгкKaijuЃЈ38%ЃЉЁЃ
Homo sapiens reads misclassifications as bacteria and decontamination test

етЯюбаОПНвЪОСЫЕБЧАКъЛљвђзщЗжРрЙЄОпдкЛЗОГбљБОжаЕФШ§ДѓОжЯоЃК1ЃЉЪ§ОнПтИВИЧВЛШЋЕМжТCandidatusОњЪєТЉМьЃЛ2ЃЉецКЫгыдКЫађСаНЛВцЮлШОЃЈШчШЫРрЁњХБдГцЃЉЃЛ3ЃЉЖЬЖСГЄзщзАжЪСПжЦдМMAGsЗжРраЇЙћЁЃзїепНЈвщЃК1ЃЉгХЯШбЁгУKaijuЃЈmЁн30ВЮЪ§ЃЉЃЛ2ЃЉЖдСйДВбљБОВЩгУKraken2ЃЈжУаХЖШ0.05-0.3ЃЉЃЛ3ЃЉБиаыНјааШЫРрађСаШЅЮлШОЁЃИУЛљзМбаОПЮЊЗЯЫЎДІРэЮЂЩњЮязщЕФОЋзМНтЮіНЈСЂСЫЗНЗЈбЇБъзМЃЌгШЦфЖдзЪдДЛиЪеЃЈШчОлєЧЛљЭщЫсѕЅPHAЩњВњЃЉЕФОњШКЕїПиОпгаживЊжИЕММлжЕЁЃ
ЩњЮяЭЈЮЂаХЙЋжкКХ
жЊУћЦѓвЕеаЦИ
НёШеЖЏЬЌ | ШЫВХЪаГЁ | аТММЪѕзЈРИ | жаЙњПЦбЇШЫ | дЦеЙЬЈ | BioHot | дЦНВЬУжБВЅ | ЛсеЙжааФ | ЬиМлзЈРИ | ММЪѕПьбЖ | УтЗбЪдгУ
АцШЈЫљга ЩњЮяЭЈ
Copyright© eBiotrade.com, All Rights Reserved
СЊЯЕаХЯфЃК
дСICPБИ09063491КХ
