
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
多巴胺受体拮抗剂的给药途径依赖性药代动力学:基于可卡因与阿扑吗啡大鼠自我给药模型的实时生物测定
【字体: 大 中 小 】 时间:2025年07月07日 来源:Scientific Reports 3.8
编辑推荐:
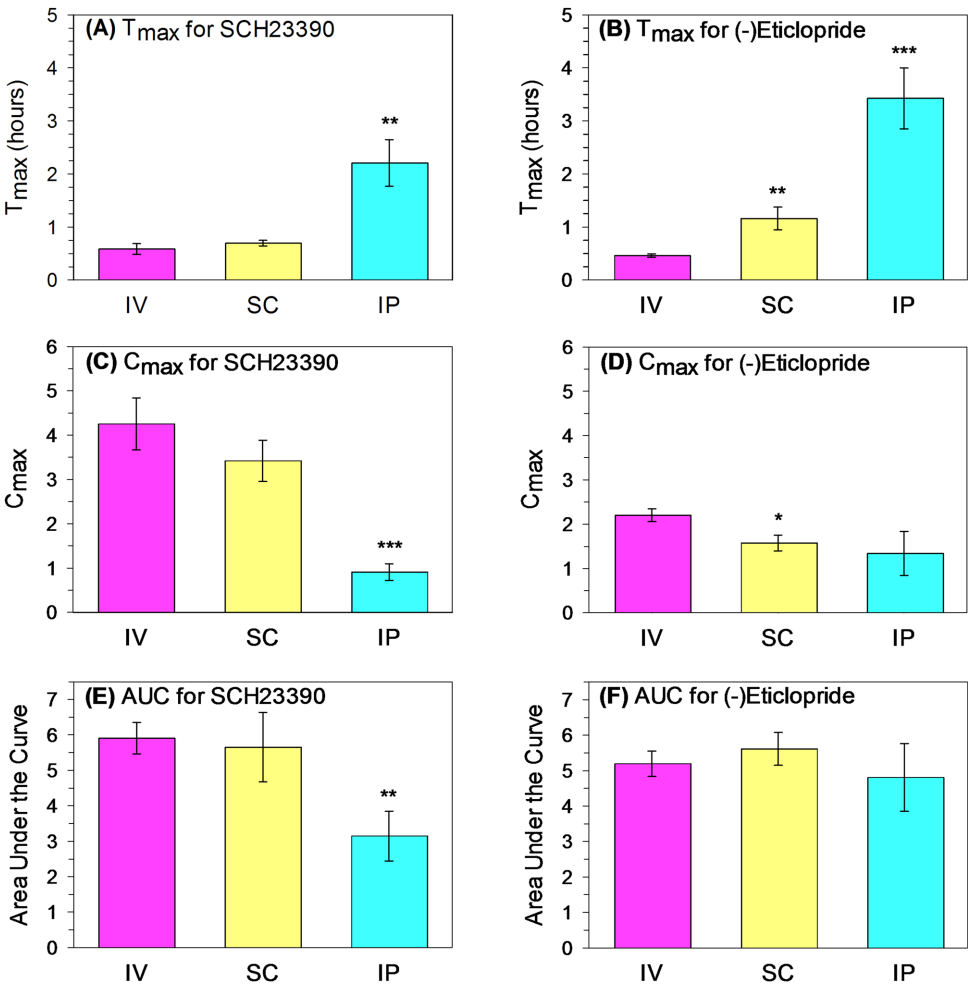
本研究针对竞争性多巴胺受体拮抗剂的药代动力学(PK)与药效学(PD)相互干扰的难题,利用可卡因(cocaine)和阿扑吗啡(apomorphine)大鼠自我给药行为模型,定量分析SCH23390(D1拮抗剂)与(-)Eticlopride(D2拮抗剂)经静脉(IV)、皮下(SC)和腹腔(IP)给药后的时效关系。结果表明,不同给药途径显著影响拮抗剂起效时间(Tmax)与峰效应(Cmax),但曲线下面积(AUC)相似,证实该模型可作为实时监测脑内拮抗剂PK/PD的活体生物测定系统,为中枢药物研发提供新方法。
在药物成瘾研究中,可卡因自我给药行为被解释为一种独特的药代动力学/药效学(PK/PD)模型:动物仅在血药浓度高于"启动阈值"且低于"饱足阈值"的"强迫区"内持续按压杠杆。这一现象背后,竞争性多巴胺受体拮抗剂可通过升高饱足阈值加速自我给药行为,但其时效曲线既反映药代动力学过程(如吸收速率),又受药效学特性(如受体亲和力)干扰,导致传统方法难以精确解析拮抗剂的脑内动态过程。
为解决这一难题,美国辛辛那提大学医学院的研究团队创新性地将可卡因(间接多巴胺激动剂)和阿扑吗啡(直接激动剂)的大鼠自我给药行为发展为实时生物测定系统。通过对比D1拮抗剂SCH23390和D2拮抗剂(-)Eticlopride经静脉(IV)、皮下(SC)和腹腔(IP)三种途径给药后对自我给药行为的调控规律,研究者首次实现了拮抗剂脑内药代动力学的活体动态监测。相关成果发表于《Scientific Reports》。
拮抗剂显著改变自我给药模式
注射0.02 μmol/kg (-)Eticlopride后,阿扑吗啡自我给药间隔缩短(即速率加快),伴随计算血药浓度上升:

给药途径决定PK特征
浓度比-1(反映拮抗剂效应强度)分析显示:

 生物通微信公众号
生物通微信公众号
知名企业招聘

