
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
综述:HPV阳性宫颈癌与口咽癌分子机制及临床差异:一项批判性叙述综述
【字体: 大 中 小 】 时间:2025年07月08日 来源:BMC Medicine 7.7
编辑推荐:
这篇综述深入探讨了HPV(人类乳头瘤病毒)阳性宫颈鳞状细胞癌(CSCC)与口咽鳞状细胞癌(OPSCC)的分子机制差异,重点解析了E6/E7致癌蛋白如何通过破坏p53/RB通路、诱导基因组不稳定性和免疫逃逸驱动肿瘤发生,并揭示了二者对放疗敏感性的显著差异(OPSCC高敏感性 vs CSCC无优势)。文章系统比较了病毒DNA整合模式、DNA修复(HR/NHEJ)失调及组织特异性微环境的影响,为HPV相关癌症的精准治疗提供了新视角。
HPV(人类乳头瘤病毒)导致全球5%的癌症负担,其中高危型HPV-16和HPV-18与70%的OPSCC和几乎所有CSCC相关。尽管两种癌症共享病毒病因,其临床行为却截然不同:HPV阳性OPSCC表现出显著的放射敏感性,允许治疗减量并改善预后,而HPV阳性CSCC仍需采用与HPV阴性病例相同的激进治疗方案。这种差异促使研究者探索分子层面的驱动因素。
HPV的8-kbp环状基因组分为早期(E)、晚期(L)和非编码上游调控区(URR)。E区编码E6/E7等致癌蛋白,L区编码衣壳蛋白L1/L2。病毒通过基底上皮细胞的微损伤感染,依赖宿主复制机制完成生命周期。E2蛋白启动病毒DNA复制并与宿主有丝分裂染色体结合,确保病毒基因组在细胞分裂中稳定传递。E6/E7通过降解p53和RB解除细胞周期调控,同时激活MAPK通路,最终导致细胞无限增殖和分化阻滞。
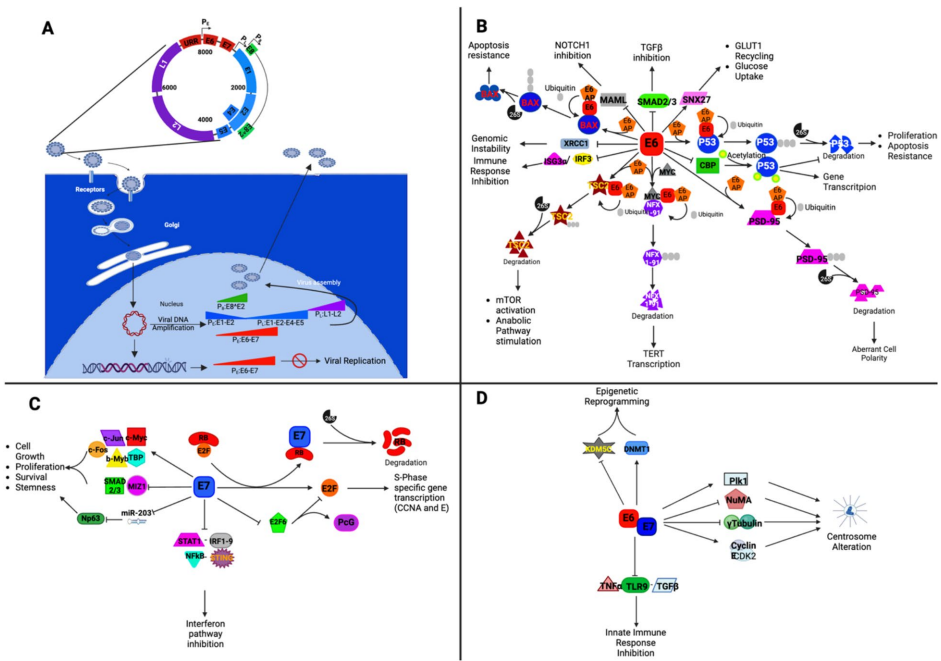
E6通过E6-AP泛素连接酶促使p53降解,同时抑制干扰素通路(如IRF-3)和端粒酶激活(通过NFX1-91降解)。E7则靶向RB蛋白,释放E2F转录因子,驱动细胞周期进程。二者还共同破坏DNA修复机制:E6结合XRCC1削弱单链断裂修复,E7通过TGF-β/miR-182轴抑制同源重组(HR),导致基因组不稳定性加剧。
70-85%的HPV阳性癌症存在病毒DNA整合,通常发生在宿主基因组脆弱位点。整合导致E2 ORF破坏,解除对E6/E7的转录抑制,形成病毒-宿主融合转录本。CSCC中常见整合基因包括FHIT和HMGA2,而OPSCC更易影响RAD51B和免疫相关基因(如PD-L1),这种差异可能解释二者不同的免疫微环境和治疗响应。
HPV阳性OPSCC的放射敏感性可能源于:
目前,HPV阳性OPSCC已实现放疗剂量从70 Gy降至45-60 Gy而不影响生存,而CSCC仍需80-90 Gy高剂量联合近距离放疗。未来需通过HPV整合位点分析和分子分型(如免疫型vs角质化型)进一步个体化治疗策略,同时探索放疗与靶向E6/E7或免疫治疗的联合方案。
HPV驱动的癌症虽共享致癌机制,但组织特异性因素(如干细胞微环境、宿主基因突变谱)导致显著的临床异质性。破解这些差异将优化治疗策略,减少毒性并改善预后。
生物通微信公众号
知名企业招聘

