
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
PHF21A错义变异c.1285G>A致神经发育障碍的分子机制:剪接缺陷导致基因剂量不足而非DNA结合功能障碍
【字体: 大 中 小 】 时间:2025年07月08日 来源:Cellular and Molecular Neurobiology 3.6
编辑推荐:
本研究针对PHF21A相关神经发育障碍(IDDBCS)中唯一报道的错义变异c.1285G>A开展机制解析。通过系统性分析该变异对mRNA剪接及DNA结合功能的影响,发现其通过破坏U1 snRNA识别位点显著降低PHF21A剪接效率(神经元和非神经元细胞分别下降55%和63%),导致内含子滞留和外显子跳跃,引发无义介导衰变(NMD)和基因剂量不足。意外的是,该变异未影响AT-hook基序的DNA结合能力。此发现不仅阐明IDDBCS的致病新机制,更为靶向剪接修复的精准治疗提供理论依据。
在人类基因组中,PHF21A作为组蛋白修饰的"解读器",通过其PHD结构域特异性识别未甲基化的H3K4(H3K4me0),并通过AT-hook基序结合DNA,在神经发育中扮演关键角色。该基因杂合性缺失会导致智力障碍、行为异常和颅面畸形综合征(IDDBCS)。有趣的是,文献中唯一报告的致病错义变异c.1285G>A位于第13号外显子末端――这个特殊位置如同"分子十字路口":既是AT-hook核心氨基酸密码(Gly429),又毗邻神经元特异性微外显子(nE14),可能同时影响蛋白质功能和RNA剪接。这为科学家提供了破解PHF21A致病机制的绝佳契机。
密歇根大学医学院的研究团队构建了涵盖变异位点的PHF21A微型基因(minigene)系统,分别在293T细胞(非神经元)和原代小鼠皮层神经元中模拟剪接过程。通过RT-qPCR定量剪接效率,结合Illumina MiSeq深度解析剪接异常模式,并在患者来源淋巴母细胞中验证结果。为评估变异对蛋白质功能的影响,团队表达纯化野生型与突变型PHF21A的AT-hook/PHD片段,采用凝胶迁移实验(EMSA)和生物膜层干涉技术(BLI)定量分析DNA结合能力。
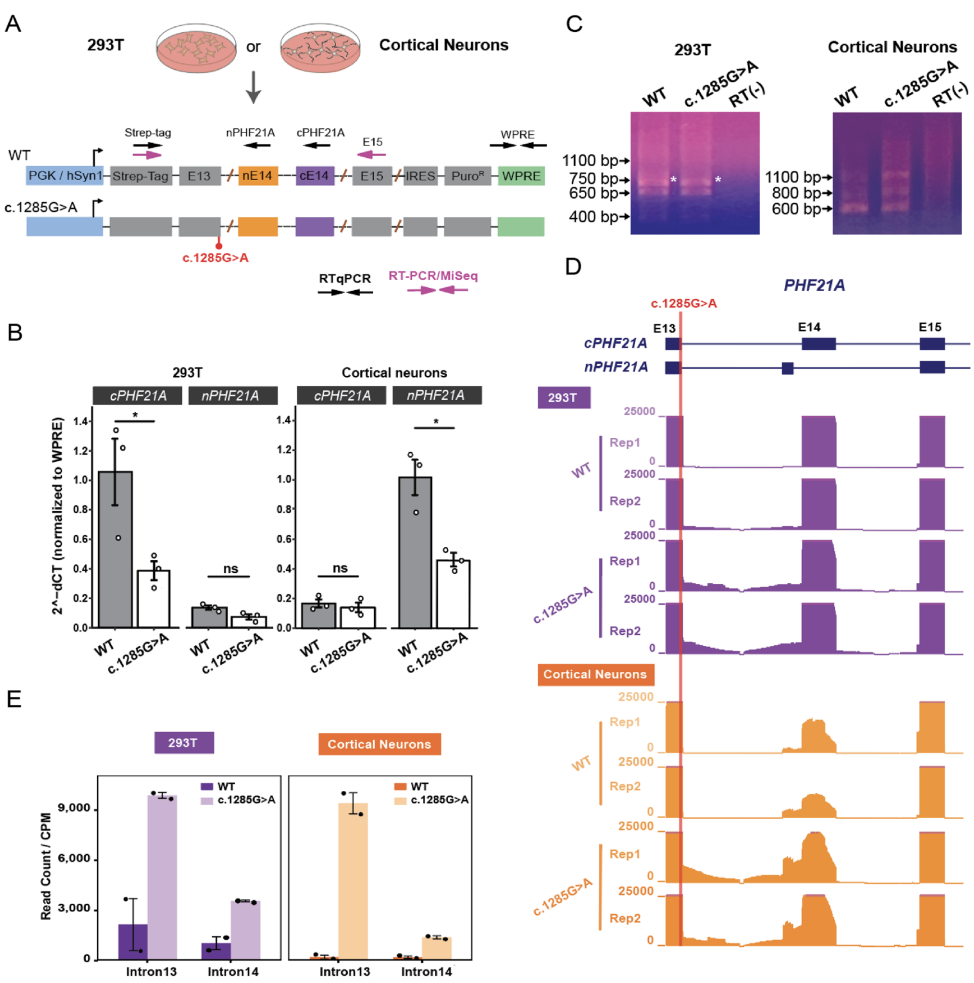

本研究首次阐明c.1285G>A致病的"双通道机制":
值得注意的是,神经元特异性剪接调控元件在minigene系统中未能完全重现,提示染色质环境对PHF21A剪接调控的重要性。这为后续研究指明方向:需在基因组原位或类器官模型中进一步验证剪接调控网络。
PHF21A c.1285G>A通过破坏剪接位点而非损害DNA结合功能导致神经发育障碍,这一发现不仅解决IDDBCS致病机制的长期争议,更开创了靶向RNA剪接的治疗新策略。该研究凸显了整合剪接分析与功能验证在精准医疗中的重要性――有时基因变异的"位置密码"比氨基酸改变本身更具临床意义。
生物通微信公众号
知名企业招聘

