
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
综述:高海拔脑水肿形成机制的研究进展
【字体: 大 中 小 】 时间:2025年07月09日 来源:Discover Medicine
编辑推荐:
这篇综述系统梳理了高海拔脑水肿(HACE)的多因素发病机制,重点探讨了低氧环境(HH)下血脑屏障(BBB)破坏、小胶质细胞过度激活、氧化应激(ROS)与炎症(NF-κB/IL-1β)的协同作用,以及线粒体功能障碍(Drp1)、离子失衡(Na+/K+-ATPase)和AQP4异常等关键病理环节,为HACE的靶向干预提供了新思路。
高海拔脑水肿(HACE)是急性高原病(AMS)的危重阶段,其发病机制涉及低氧环境触发的级联反应。随着人类高原活动增加,HACE发病率上升,表现为共济失调、意识障碍等症状,严重威胁健康。尽管高原医学研究取得进展,但HACE的分子机制尚未完全阐明,动物模型与人类病理差异(如病程缩短、遗传同质性)进一步增加了研究难度。
血脑屏障(BBB)由内皮细胞、基底膜和星形胶质细胞终足构成,是维持脑内环境稳定的关键屏障。低氧条件下,缺氧诱导因子(HIFs)上调血管内皮生长因子(VEGF),导致紧密连接蛋白(Claudin-5/Occludin)减少,破坏BBB完整性。

研究发现,核呼吸因子1(NRF1)通过上调小窝蛋白1(CAV-1)促进Claudin-5的内吞和自噬降解,加剧BBB通透性。此外,低氧激活NF-κB/NLRP3炎症小体,释放IL-1β等促炎因子,协同MMP-9降解基底膜,共同驱动血管源性脑水肿。
低氧环境下,线粒体电子传递链异常导致ROS爆发,12/15-脂氧合酶活性改变诱导线粒体嵴结构损伤,细胞色素c释放激活神经元凋亡。研究显示,线粒体分裂抑制剂(mdivi-1)通过抑制Drp1磷酸化,减少NF-κB介导的TNF-α/IL-6释放,缓解脑水肿。

抗氧化酶(SOD/CAT)活性下降和丙二醛(MDA)积累进一步加剧氧化损伤。自由基清除剂(如褪黑素)通过调控AQP4表达和抑制VEGF,显示出抗HACE潜力。
低氧导致Na+/K+-ATPase功能障碍,引发细胞内Na+蓄积和K+外流,早期以细胞毒性水肿为主;随着缺氧时间延长,Cl-通道(CFTR/VRAC)和TRPV1钙通道激活,推动离子性水肿形成。钙超载通过激活蛋白酶级联反应,加重神经元死亡。
低氧促使小胶质细胞向促炎M1型极化,释放TNF-α等因子;而M2型则发挥神经保护作用。乳酸代谢异常通过HIF-1α/LDH-A轴诱导细胞外酸化,激活TRPV1和HDAC抑制,形成“代谢-炎症”恶性循环。
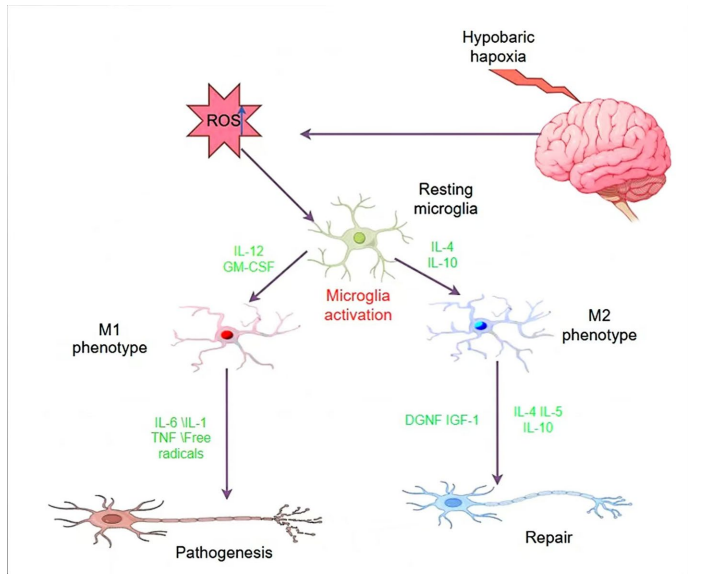
当前研究存在样本区域性偏倚、长程机制不明等问题。靶向CAV-1/Claudin-5内吞、Drp1介导的线粒体分裂、以及乳酸代谢调控(如LDH-A抑制剂)可能是突破方向。多中心临床研究和跨机制交互作用(如离子通道-AQP4耦合)需进一步探索。
生物通微信公众号
知名企业招聘

