
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
基因组溯源技术揭示中国消除疟疾后一例无旅行史恶性疟原虫病例的传播链
【字体: 大 中 小 】 时间:2025年07月12日 来源:Malaria Journal 2.4
编辑推荐:
本研究针对中国消除疟疾后出现的无明确旅行史恶性疟原虫(Plasmodium falciparum)感染病例,采用全基因组测序(WGS)、主成分分析(PCA)和血缘一致性(IBD)等基因组溯源技术,成功追踪到该病例与刚果民主共和国(DRC)输入病例的遗传关联(IBD=0.9),证实其为输入性传播而非本地感染。该研究为消除后阶段的疟疾监测提供了高精度溯源工具,对维持中国无疟状态具有重要意义。
在全球疟疾防控取得重大进展的背景下,中国于2021年获得世界卫生组织(WHO)的疟疾消除认证。然而这个胜利并非一劳永逸――随着国际人员流动增加,输入性疟疾病例持续构成疫情复燃风险。2019年重庆报告的一例特殊病例敲响了警钟:患者无出境史却确诊恶性疟原虫感染,这究竟是监测漏洞还是本地传播死灰复燃?传统流行病学调查陷入僵局时,基因组学技术为破解谜题带来了曙光。
中国疾病预防控制中心寄生虫病预防控制所(国家热带病研究中心)的研究团队创新性地将分子溯源技术应用于消除后监测。通过对该病例和同期输入病例的恶性疟原虫进行全基因组测序,结合生物信息学分析,最终在《Malaria Journal》发表了这项具有里程碑意义的研究。
研究采用三大关键技术:全基因组测序(WGS)获取重庆病例及非洲参考菌株的基因组数据;主成分分析(PCA)和邻接树(Neighbor-Joining tree)进行地理溯源;血缘一致性(IBD)分析评估样本间遗传关联。所有样本均来自2015-2019年重庆报告的输入病例及中国-缅甸边境对照株。
主要结果
PCA与系统发育分析定位感染源
基因组数据清晰显示:神秘病例的疟原虫聚于中非分支,与刚果(DRC)输入株遗传距离最近。而中国-缅甸边境株则独立成簇,验证了分析方法的特异性。
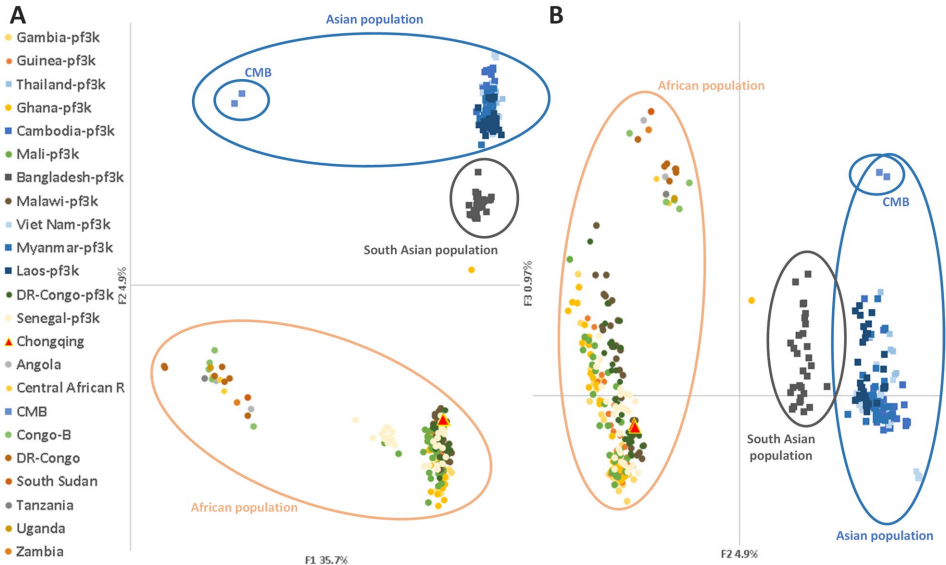
IBD分析揭示传播链
关键发现来自血缘分析――该病例与同期住院的DRC输入病例存在惊人遗传相似性(IBD=0.9),远超随机匹配水平。这种亲缘关系强烈暗示医院内交叉感染可能,尽管传统调查未发现明确接触史。

讨论与意义
这项研究首次将基因组流行病学应用于中国消除后阶段的疟疾监测,突破了三重技术壁垒:在低寄生虫密度样本中获取高质量基因组数据;建立非洲疟原虫的遗传地理图谱;开发适用于消除环境的溯源流程。Fei Luo等学者证实,即便在传统调查"山穷水尽"时,WGS-PCA-IBD技术组合仍能精准锁定感染源。
该案例凸显消除后阶段的特殊挑战:输入病例可能通过非典型途径(如医疗环境暴露)引发"隐蔽传播"。研究团队建议将基因组工具整合至"1-3-7"监测策略,在病例核实阶段增加分子溯源模块。这不仅可识别高风险传播场景,更能为全球消除疟疾提供技术范本――正如作者强调:"基因组学是维持无疟状态的‘基因显微镜’"。
这项研究标志着中国疟疾防控从"清零"迈向"防复燃"的新阶段。随着测序成本降低和分析流程标准化,基因组监测有望成为全球消除疟疾的标配工具,为最终实现 eradication(根除)目标提供关键技术支撑。
生物通微信公众号
知名企业招聘

