
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
综述:铁蛋白自噬与铁死亡在缺血性脑卒中中的交互作用:调控机制与治疗意义
【字体: 大 中 小 】 时间:2025年07月21日 来源:Cellular and Molecular Neurobiology 3.6
编辑推荐:
这篇综述系统阐述了缺血性脑卒中(Ischemic Stroke)中铁死亡(Ferroptosis)和铁蛋白自噬(Ferritinophagy)的分子机制及其交互作用。文章揭示了铁代谢紊乱(TfR1/DMT1/FPN1)、脂质过氧化(ACSL4/LOXs)和抗氧化系统(xCT/GSH/GPX4)失衡的核心病理过程,并探讨了靶向NCOA4-FTH1轴和cGAS-STING通路的新型治疗策略,为缺血性脑损伤的干预提供了新视角。
铁死亡与铁蛋白自噬:缺血性脑卒中的双重杀手
铁死亡――脑缺血的新型细胞死亡方式
铁死亡是2012年发现的一种铁依赖性程序性细胞死亡形式,其特征是铁过载、谷胱甘肽(GSH)耗竭和脂质过氧化物积累。在缺血性脑卒中中,铁代谢紊乱表现为转铁蛋白受体1(TfR1)和二价金属转运体1(DMT1)上调,而铁转运蛋白(FPN1)下调,导致神经元内游离铁水平异常升高。这些游离铁通过Fenton反应产生大量活性氧(ROS),攻击细胞膜上的多不饱和脂肪酸(PUFAs),在长链脂酰辅酶A合成酶4(ACSL4)和15-脂氧合酶(15-LOX)的催化下形成致命的脂质过氧化物。
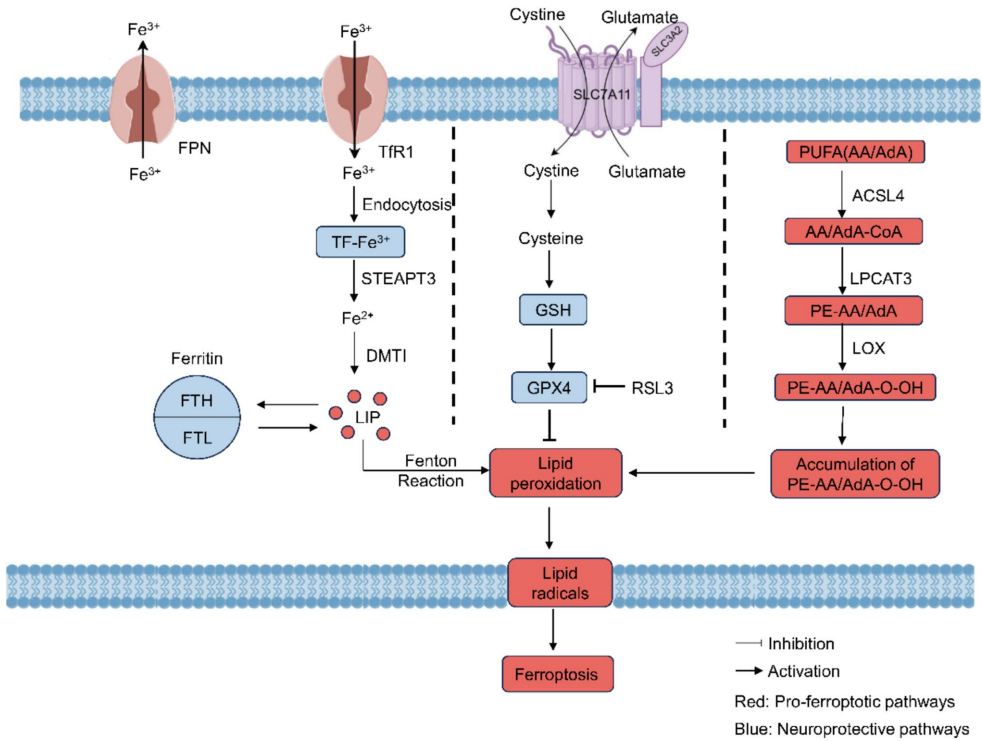
抗氧化防御系统的崩溃
半胱氨酸/谷氨酸逆向转运体(System Xc-)和谷胱甘肽过氧化物酶4(GPX4)构成关键的抗氧化防线。脑缺血时,谷氨酸兴奋毒性抑制System Xc-活性,导致半胱氨酸摄取减少、GSH合成受阻。GPX4失活后,无法还原脂质过氧化物,最终引发细胞膜破裂。值得注意的是,转录因子Nrf2可通过激活抗氧化反应元件(ARE)上调GPX4和SLC7A11表达,而p53则通过SAT1/ALOX15轴促进脂质过氧化,二者在铁死亡调控中扮演着"阴阳"角色。
铁蛋白自噬――铁死亡的催化剂
铁蛋白自噬是由核受体辅激活因子4(NCOA4)介导的选择性自噬过程。在生理状态下,NCOA4通过其C端结构域与铁蛋白重链(FTH1)的R23位点结合,形成复合体后被溶酶体降解释放铁离子。脑缺血再灌注损伤(CIRI)中,cGAS-STING通路异常激活导致NCOA4积累,引发铁蛋白过度降解。释放的铁离子进一步加剧氧化应激,形成恶性循环。研究发现,抑制USP14介导的NCOA4去泛素化或使用银杏内酯B阻断NCOA4-FTH1相互作用,均可显著减轻神经元损伤。

神经胶质细胞的推波助澜
小胶质细胞通过TfR1/DMT1途径摄取过量铁离子,并通过cGAS-STING-NLRP3炎症小体轴促进M1型极化,分泌IL-1β等促炎因子抑制GPX4活性。星形胶质细胞则通过外泌体将铁相关蛋白传递给神经元,加剧铁死亡进程。有趣的是,少突胶质前体细胞在缺血后通过铁依赖性机制促进髓鞘再生,提示不同胶质细胞在铁代谢中具有时空特异性。
治疗策略与挑战
目前靶向铁死亡的治疗手段包括:铁螯合剂去铁胺(DFO)、GPX4激活剂硒代半胱氨酸、ALOX抑制剂齐留通等。最新研究显示,电针刺激通过调控NCOA4-FTH1轴改善神经功能,而携带miR-760-3p的外泌体可靶向抑制CHAC1表达。然而,血脑屏障(BBB)穿透性差和患者个体差异仍是临床转化的主要障碍。未来研究需开发新型递药系统,并探索铁蛋白自噬与线粒体自噬(Mitophagy)的交互机制。
生物通微信公众号
知名企业招聘

