
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
综述:癌症中坏死性凋亡的表观遗传、转录后和翻译后修饰调控机制
《Journal of Hematology & Oncology》:Necroptosis in cancer: insight from epigenetic, post-transcriptional and post-translational modifications
【字体: 大 中 小 】 时间:2025年07月31日 来源:Journal of Hematology & Oncology 40.4
编辑推荐:
这篇综述系统阐述了坏死性凋亡(necroptosis)在癌症中的双重作用及其调控机制。作者深入分析了表观遗传修饰(DNA甲基化、组蛋白修饰)、转录后调控(非编码RNA、可变剪接)和翻译后修饰(磷酸化、泛素化等)如何通过调控RIPK1/RIPK3/MLKL信号轴影响肿瘤免疫微环境,为开发新型抗癌靶向治疗提供了理论依据。特别强调了通过修饰干预诱导免疫原性细胞死亡(ICD)的治疗潜力。
坏死性凋亡作为一种兼具坏死和凋亡特征的程序性细胞死亡形式,其核心调控机制依赖于RIPK1-RIPK3-MLKL信号轴的级联激活。当死亡受体如TNFR1、TLR4等被相应配体激活后,RIPK1通过其死亡结构域和RHIM结构域介导信号转导。在caspase-8活性受抑制时,RIPK1与RIPK3形成复合体IIb(necrosome),进而磷酸化MLKL导致其寡聚化和膜穿孔,最终释放损伤相关分子模式(DAMPs)引发炎症反应。
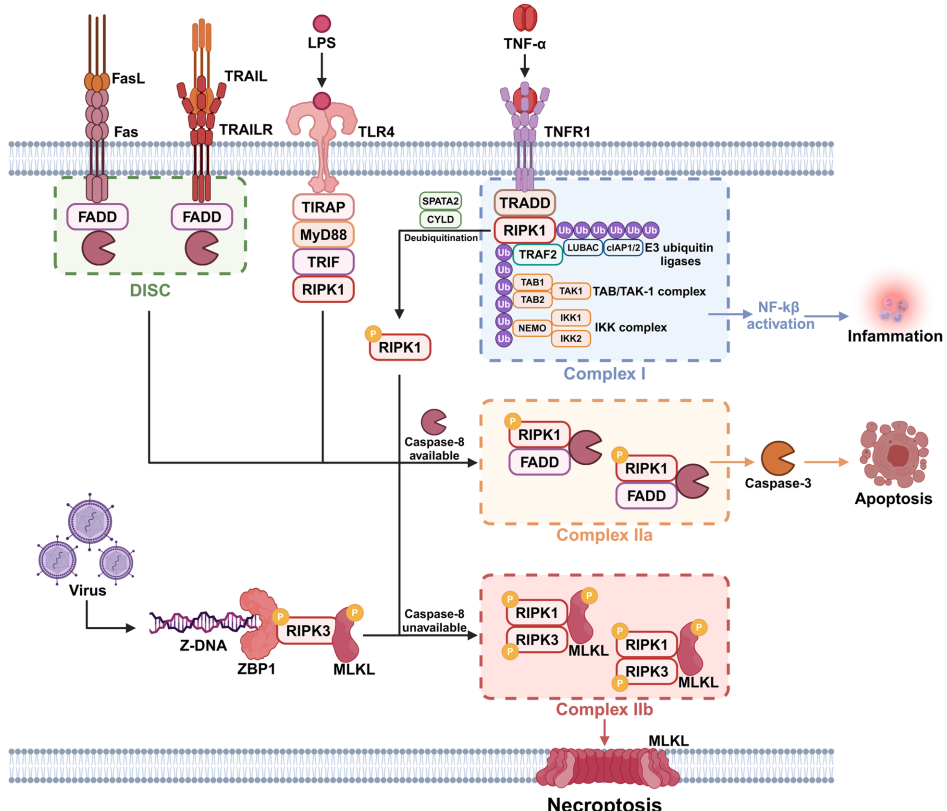
DNA甲基化通过DNMTs调控关键基因表达:在CRC中,IL-6/STAT3通路通过上调DNMT1/3B导致TNF-α启动子超甲基化,抑制MDSCs的坏死性凋亡;而在恶性间皮瘤和AML中,RIPK3启动子甲基化导致化疗抵抗。组蛋白修饰同样发挥重要作用:G9a介导的H3K9me2沉默修饰抑制乳腺癌中TNF-α表达,而复发乳腺癌中RIPK3启动子区激活型修饰(H3K4me3/H3K9Ac)使其对半胱氨酸剥夺敏感。

miR-675通过lncRNA H19来源促进肝癌中FADD下调,促使死亡方式向坏死性凋亡转换;而miR-204-3p则通过抑制RIPK1/MLKL表达阻碍胃癌细胞坏死性凋亡。可变剪接产生MLKL不同亚型:促坏死型mMLKL2通过C端α螺旋插入疏水沟激活死亡信号,而mMLKL1因插入8肽序列阻碍该过程。
磷酸化修饰形成精密调控网络:RIPK1 S166自磷酸化是激活标志,而IKK介导的S25磷酸化则抑制其细胞毒性;RIPK3 S227被CK1或自身磷酸化后招募MLKL,而Ppm1b通过去磷酸化该位点抑制坏死性凋亡。泛素化修饰呈现双向调控:PELI1介导RIPK1 K115位K63连接泛素化促进坏死小体形成,而CHIP通过K48连接泛素化导致RIPK3溶酶体降解。

靶向表观遗传的药物如地西他滨(DNMT抑制剂)和BIX-01294(G9a抑制剂)可重新激活RIPK3表达;而靶向翻译后修饰的化合物如Fostamatinib(TAK1激活剂)和PX-12(Trx-1抑制剂)分别通过调控磷酸化和二硫键形成促进坏死性凋亡。纳米载体递送shikonin等天然化合物可特异性诱导肿瘤细胞坏死性凋亡,同时激活cGAS-STING通路增强抗PD-1疗效,为克服免疫治疗耐药提供新策略。

该综述系统整合了多维度调控机制,揭示通过精确干预坏死性凋亡通路可同时实现肿瘤细胞杀伤和免疫微环境重塑,为发展"表观遗传-免疫"联合疗法提供了重要理论基础。未来研究需进一步阐明不同修饰间的交叉对话,以及如何平衡其促癌与抑癌双重效应。
生物通微信公众号

