
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
SARS-CoV-2 NSP5蛋白酶P132H突变通过阻断MAVS降解缓解其对干扰素-β激活的抑制作用
《Cellular and Molecular Life Sciences》:The P132H mutation of SARS-CoV-2 NSP5 relieves its inhibition on interferon-β activation via blocking MAVS degradation
【字体: 大 中 小 】 时间:2025年08月01日 来源:Cellular and Molecular Life Sciences 6.2
编辑推荐:
本研究针对SARS-CoV-2奥密克戎变异株致病性降低的机制展开探索,发现其NSP5蛋白的P132H突变通过削弱与E2泛素结合酶UbcH5b的相互作用,阻断MAVS蛋白K136位点的K63泛素化降解,从而恢复被抑制的IFN-β信号通路活性。该研究揭示了非刺突蛋白突变在病毒免疫逃逸中的关键作用,为理解奥密克戎低致病性提供了新视角。
新冠病毒奥密克戎变异株的致病性为何显著低于早期毒株?这一科学谜题背后可能隐藏着非刺突蛋白的关键作用。湖南大学医学病毒学湖南省重点实验室的研究团队将目光聚焦于病毒编码的NSP5蛋白酶――这个在冠状病毒复制中扮演核心角色的"分子剪刀"竟还具有抑制宿主先天免疫的"副业"。研究发现,奥密克戎特有的P132H突变像一把钝化的剪刀,虽然保留了切割病毒多蛋白的基本功能,却意外失去了压制宿主免疫系统的"暗黑技能"。
研究人员通过构建P132H突变病毒株,发现该突变显著降低病毒复制效率并增强干扰素应答。深入机制研究表明,野生型NSP5作为E3泛素连接酶,会招募UbcH5b对MAVS蛋白K136位点进行K63泛素化修饰,进而促使其通过蛋白酶体降解。而P132H突变通过破坏NSP5与UbcH5b之间的氢键网络,削弱了这种抑制作用,使MAVS得以激活下游干扰素信号。这项发表于《Cellular and Molecular Life Sciences》的研究,首次揭示了非结构蛋白突变在奥密克戎低致病性中的关键作用。
研究主要采用以下技术方法:1) 构建WH-NSP5-P132H突变病毒株进行体外感染实验;2) 双荧光素酶报告系统检测IFN-β启动子活性;3) 免疫共沉淀结合免疫印迹分析蛋白互作;4) 环己酰亚胺追踪实验测定蛋白半衰期;5) 计算机模拟预测蛋白互作界面。
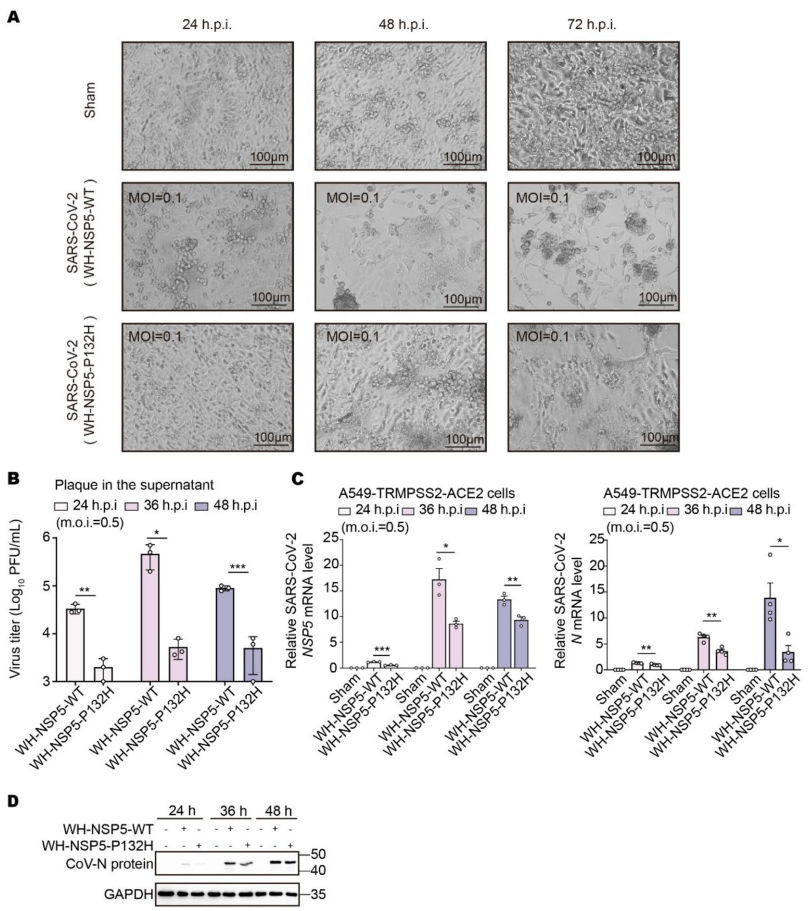
P132H突变降低SARS-CoV-2复制能力
通过构建武汉株背景的P132H突变病毒发现,该突变显著减小合胞体规模,降低病毒滴度。qRT-PCR和免疫印迹显示病毒N蛋白和NSP5表达量下降,证实该突变减弱了病毒复制能力。
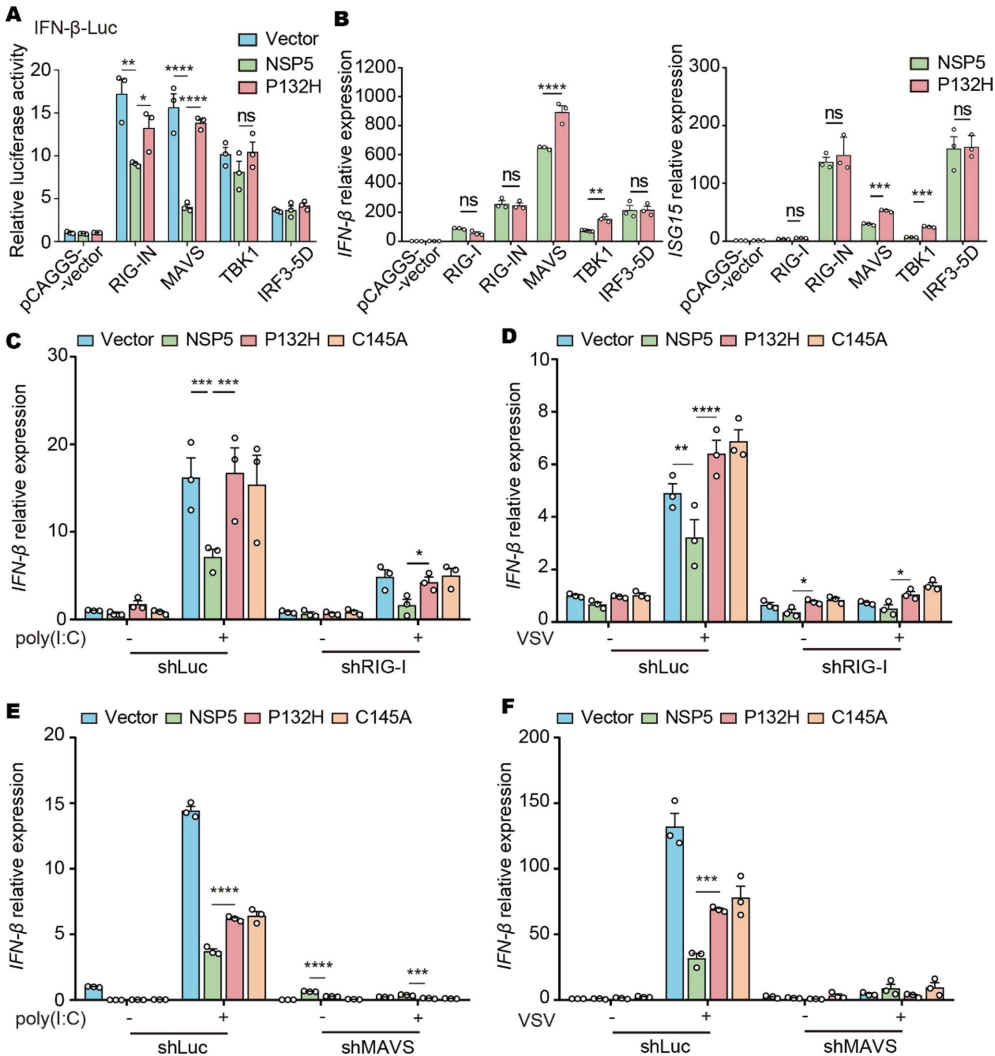
P132H削弱NSP5对I型干扰素的抑制
转录组分析显示P132H影响RIG-I样受体信号通路。病毒感染实验证实突变株能更强诱导IFN-β及其下游ISG15、CXCL10等基因表达。双荧光素酶报告系统证明P132H可恢复被NSP5抑制的IFN-β启动子活性。
P132H通过靶向MAVS发挥作用
通过过表达和敲低实验锁定MAVS为关键靶点。在MAVS敲除细胞中,P132H失去对IFN-β的激活作用,而在RIG-I敲除细胞中仍保持活性。蛋白酶活性实验显示P132H不影响NSP5对病毒多蛋白的切割功能。
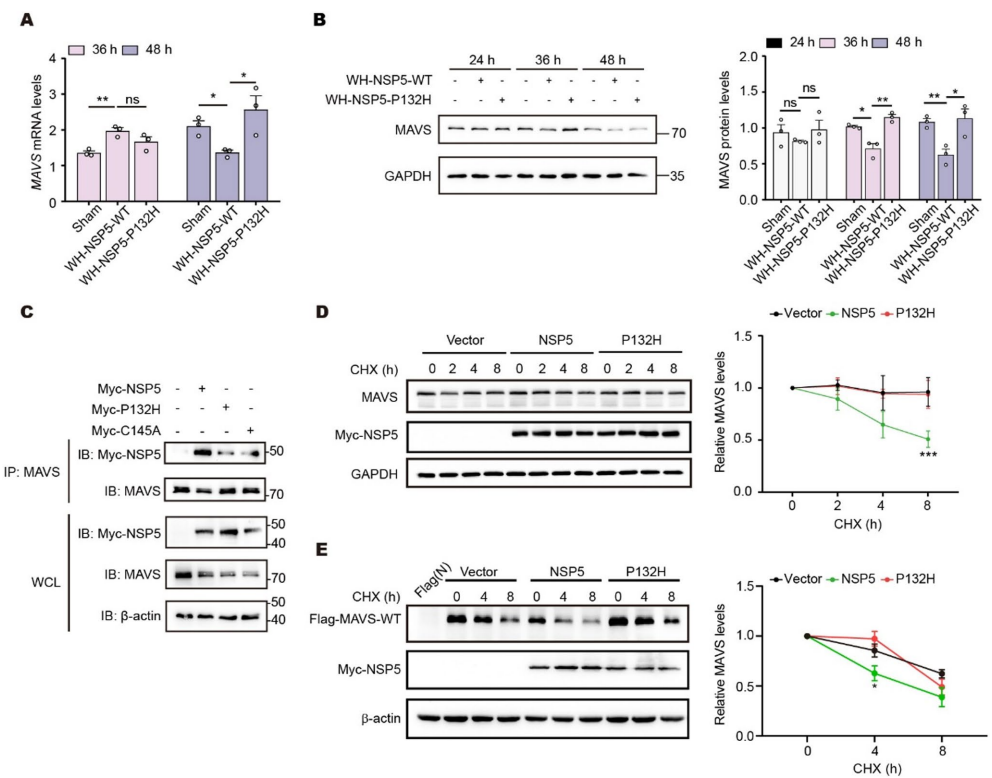
P132H阻断MAVS降解
免疫共沉淀证实NSP5与MAVS直接互作。环己酰亚胺追踪实验显示P132H延长MAVS半衰期。泛素化分析揭示NSP5促进MAVS的K63泛素化,而P132H减弱这一修饰。K136R突变体对NSP5介导的降解产生抵抗。
结构基础与机制解析
计算机模拟显示P132H破坏NSP5与UbcH5b间的氢键网络,特别是R4-D956和K236-V26/G27/D28的相互作用。免疫共沉淀证实P132H减弱NSP5与UbcH5b的结合能力,从而影响其E3泛素连接酶功能。
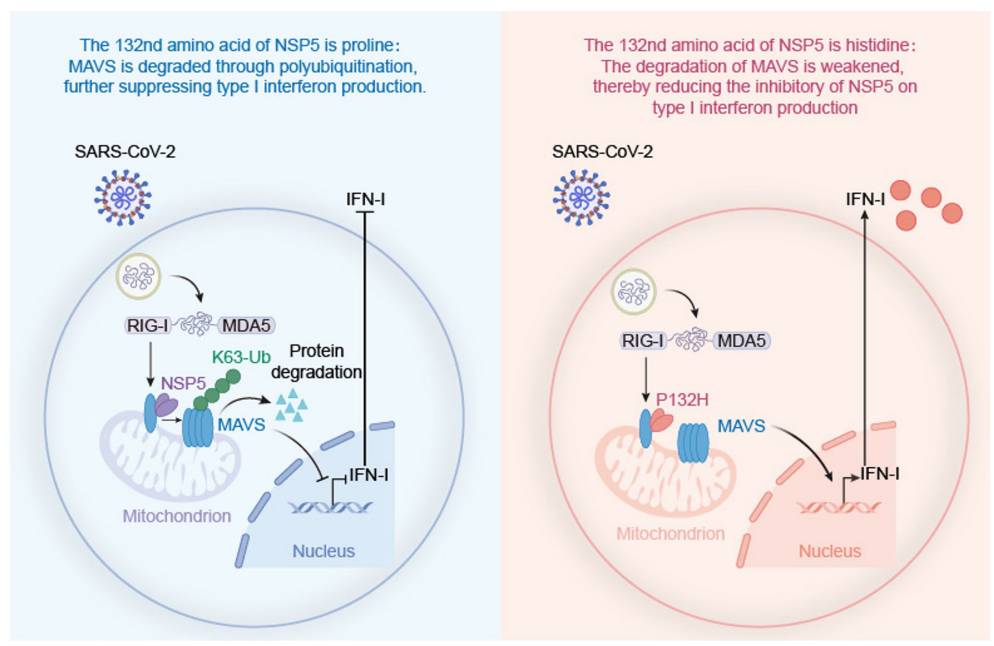
该研究系统阐明了奥密克戎NSP5-P132H突变通过"双刃剑"机制降低病毒毒力的分子基础:一方面维持病毒复制必需的蛋白酶活性,另一方面削弱免疫抑制功能。这一发现不仅解释了奥密克戎临床致病性降低的现象,更揭示了病毒非结构蛋白与宿主免疫系统的精细博弈,为未来广谱抗冠状病毒药物设计提供了新靶点。特别值得注意的是,P132H在奥密克戎所有亚系中保持>99.8%的保守性,暗示这种"自废武功"的突变可能给病毒带来其他进化优势,这一发现为理解病毒适应性进化提供了新的思考维度。
生物通微信公众号

