
-

ЩњЮяЭЈЙйЮЂ
ХуФузЅзЁЩњУќПЦММ
ЬјЖЏЕФТіВЋ
ЩњЮяЭЈЙйЮЂ
ХуФузЅзЁЩњУќПЦММ
ЬјЖЏЕФТіВЋ
ЯпСЃЬхЙІФмеЯАЧ§ЖЏBarthзлКЯеїжаадСЃЯИАћМѕЩйЕФЛњжЦбаОПЃКЛљгкЛМепЖЈжЦЛЏTafazzinЕуЭЛБфаЁЪѓФЃаЭЕФЗЂЯж
ЁОзжЬхЃК Дѓ жа аЁ ЁП ЪБМфЃК2025Фъ08дТ06Ше РДдДЃКStem Cell Reviews and Reports 4.2
БрМЭЦМіЃК
ЁЁЁЁБОбаОПеыЖдBarthзлКЯеї(BTHS)ЛМепжТУќаджаадСЃЯИАћМѕЩйЕФЛњжЦПеАзЃЌЭЈЙ§CRISPR/Cas9ЙЙНЈTAFAZZIN D75HЕуЭЛБфаЁЪѓФЃаЭЃЌЪзДЮНвЪОЯпСЃЬхФЄЕчЮЛ(ІЄІзm)ЮЩТвКЭЛюадбѕ(ROS)РлЛ§ЕМжТдьбЊзцЯИАћЙІФмЪмЫ№ЪЧКЫаФВЁРэЁЃгЁЕкАВФЩДѓбЇвНбЇдКЭХЖгЗЂЯжTazD75HаЁЪѓГЪЯжФъСфвРРЕЕФСЃЯИАћЩњГЩеЯАЃЌВЂЭЈЙ§вЦжВЪЕбщжЄЪЕИУБэаЭдДгкTAZУИЛюадШБЪЇв§ЗЂЕФЯИАћзджїадШБЯнЃЌЮЊАаЯђmPTP-CypDЭЈТЗЃЈШчЛЗцпЫиAЃЉЕФСйДВИЩдЄЬсЙЉСЫаТвРОнЁЃ
баОПБГОАгывтвх
BarthзлКЯеї(BTHS)зїЮЊвЛжжКБМћЕФXСЌЫјЯпСЃЬхМВВЁЃЌРЇШХзХШЋЧђЪ§вдЧЇМЦЕФФаадЛМепЁЃетРрЛМепВЛНіБЅЪмаФМЁВЁКЭМЁЮоСІЕФелФЅЃЌИќЪБПЬУцСйзХжТУќИаШОЕФЭўаВЁЊЁЊЦфзяП§ЛіЪзБуЪЧЗДИДЗЂзїЕФжаадСЃЯИАћМѕЩйЁЃОЁЙмСйДВЩЯГЃгУСЃЯИАћМЏТфДЬМЄвђзг(G-CSF)днЪБЛКНтжЂзДЃЌЕЋдМ47%ЕФЛМепШдЛсГіЯжжЮСЦЕжПЙЁЃЮЪЬтЕФЙиМќдкгкЃЌбЇНчЖдTAFAZZIN(TAZ)ЛљвђЭЛБфШчКЮЦЦЛЕСЃЯИАћЩњГЩЕФШЯжЊШдЪЧвЛЦЌУдЮэЁЃTAZБрТыЕФаФСзжЌ(CL)жиЙЙУИШчЭЌЯпСЃЬхЕФ"НЈжўЪІ"ЃЌЦфШБЯнЛсЕМжТЯпСЃЬхНсЙЙКЭФмСПДњаЛЕФШЋУцБРРЃЁЃШЛЖјЃЌОПОЙЪЧдьбЊИЩЯИАћЕФздЮвИќаТЪмЫ№ЃЌЛЙЪЧжаадСЃЯИАћЧАЬхЕФГЩЪьеЯАЕМжТСЫетвЛбЊвКбЇвьГЃЃПетИіУеЬтиНД§ЦЦНтЁЃ
баОПЩшМЦгыЗНЗЈ
гЁЕкАВФЩДѓбЇвНбЇдК(Indiana University School of Medicine)ЕФElizabeth A. Sierra PotchanantКЭSimon J. ConwayЭХЖгЖРБйѕшОЖЃЌВЩгУCRISPR/Cas9ЛљвђБрМММЪѕЃЌНЋвЛЮЛЕфаЭBTHSЛМепЕФTAZD75HЕуЭЛБфОЋзМв§ШыаЁЪѓЛљвђзщЃЌЙЙНЈГіШЋЧђЪзИіЮШЖЈБэДяЮоДпЛЏЛюадTAZЕААзЕФЖЏЮяФЃаЭЁЃбаОПШЫдБЭЈЙ§ЖрзщбЇЗНЗЈЯЕЭГЦРЙРСЫЭЛБфаЁЪѓЕФдьбЊЙІФмЃК
СїЪНЯИАћЪѕЗжЮідьбЊИЩЯИАћ(HSC)КЭзцЯИАћШК
МзЛљЯЫЮЌЫиМЏТфаЮГЩЪЕбщМьВтСЃЯИАћ-ОоЪЩЯИАћзцЯИАћ(GMP)ЗжЛЏЧБФм
ЙЧЫшвЦжВЪЕбщбщжЄЯИАћзджїадШБЯн
ЛМепРДдДСмАЭФИЯИАћЯпСЃЬхЙІФмМьВтЃЈJC-1КЭMitoSOXШОЩЋЃЉ
ЭИЩфЕчОЕЙлВьЯпСЃЬхГЌЮЂНсЙЙ
ЙиМќЗЂЯж
дьбЊЙІФмЕФЪБМфЬивьадЮЩТв
ЭЈЙ§БШНЯВЛЭЌжмСфаЁЪѓЕФЭтжмбЊЯѓЃЌбаОПЭХЖгВЖзНЕНвЛИіСюШЫОЊбШЕФЯжЯѓЃКTazD75HаладаЁЪѓНідкаТЩњЖљЦкКЭгзФъЦкЃЈ4жмСфЧАЃЉБэЯжГіЯджјЕФжаадСЃЯИАћМѕЩйЃЌЖјГЩФъКѓЃЈ8-10дТСфЃЉдђЛжИДе§ГЃЁЃетжжФъСфвРРЕЕФБэаЭЭъУРФЃФтСЫBTHSЛМепСйДВБэЯжЕФвьжЪадЁЃ
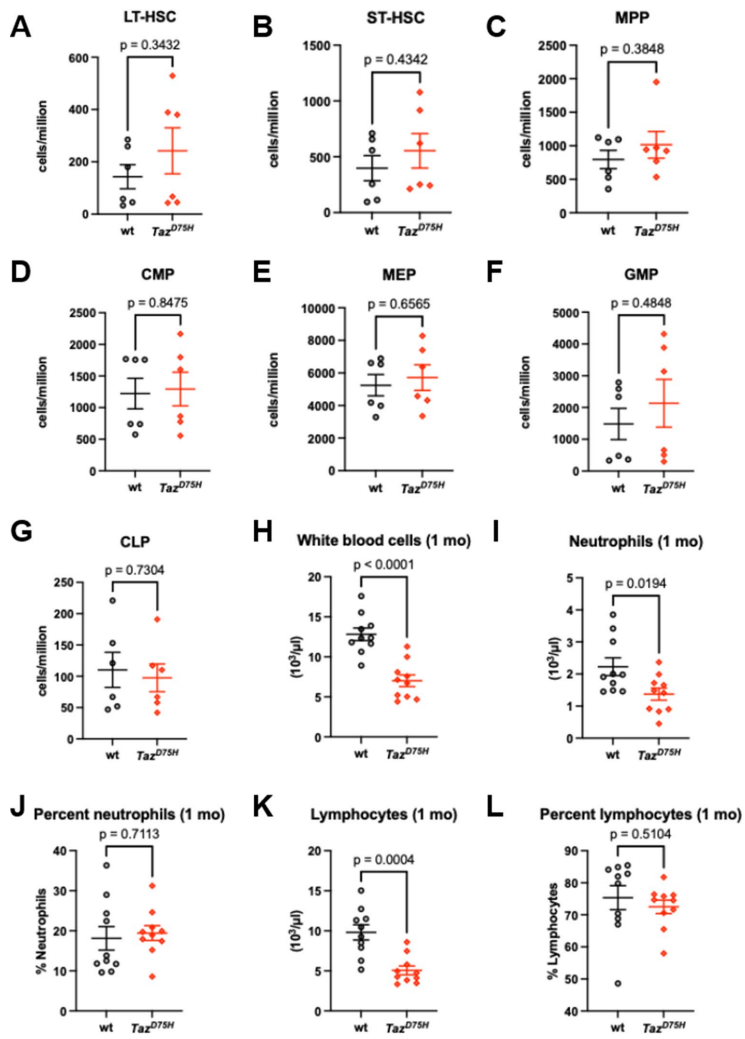
ЫшЯЕзцЯИАћЕФвўаЮШБЯн
ОЁЙмСїЪНМьВтЯдЪОИїРрдьбЊзцЯИАћЪ§СПе§ГЃЃЌЕЋМЏТфаЮГЩЪЕбщНвТЖСЫОЊШЫецЯрЃКTazD75HаЁЪѓЕФGMPЯИАћВњЩњСЃЯИАћ-ОоЪЩЯИАћМЏТф(CFU-GM)ЕФФмСІЯТНЕ40%ЁЃИќЙиМќЕФЪЧЃЌаимеЩБЩЫЪЕбщжЄЪЕетаЉзцЯИАћДІгквьГЃЕФЯИАћжмЦкЭЃжЭзДЬЌЁЊЁЊете§ЪЧСйДВЩЯG-CSFжЮСЦЗДгІВювьЕФЧБдкНтЪЭЁЃ
ЯпСЃЬхЮЃЛњЕФЗжзгжЄОн
JC-1ШОЩЋЯдЪОЭЛБфаЭдьбЊЯИАћЕФЯпСЃЬхФЄЕчЮЛ(ІЄІзm)ЦеБщНЕЕЭ30%ЃЌЖјЛМепСмАЭФИЯИАћжаMitoSOXбєадЯИАћдіМг1.5БЖЁЃЕчОЕееЦЌИќжБЙлеЙЯжСЫЯпСЃЬхжзеЭЁЂсеНсЙЙЮЩТвЕФВЁРэЬиеїЁЃЕБгУЛЗцпЫиA(CsA)зшЖЯЯпСЃЬхЭЈЭИадзЊЛЛПз(mPTP)ЪБЃЌетаЉвьГЃБЛЯджјФцзЊЃЌетЮЊАаЯђИЩдЄЬсЙЉСЫжБНгжЄОнЁЃ
СйДВЦєЪОгыеЙЭћ
етЯюбаОПЪзДЮВћУїTAZУИЛюадШБЪЇЭЈЙ§"ЯпСЃЬхЙІФмеЯА-ROSРлЛ§-зцЯИАћжмЦкзшжЭ"ЕФШ§СЊЛњжЦЕМжТBTHSбЊвКбЇвьГЃЁЃЬиБ№жЕЕУзЂвтЕФЪЧЃЌЙЧЫшвЦжВЪЕбщжЄЪЕГЩФъаЁЪѓЕФжаадСЃЯИАћМѕЩйОпгаПЩФцадЃЌетЬсЪОаТЩњЖљЦкПЩФмЪЧИЩдЄЕФЛЦН№ДАПкЁЃбаОПепЬсГіЕФCsAжЮСЦЗНАИвбдкаЁЪѓФЃаЭКЭЛМепЯИАћжаЕУЕНИХФюбщжЄЃЌЦфЭЈЙ§ЮШЖЈІЄІзmКЭМѕЩйбѕЛЏгІМЄЕФЫЋжизїгУЛњжЦЃЌгаЭћГЩЮЊG-CSFЕФЬцДњСЦЗЈЁЃ
ИУГЩЙћЕФЩюдЖвтвхЛЙдкгкЃКЮЊЦфЫћЯпСЃЬхМВВЁЯрЙиЕФбЊЯИАћМѕЩйжЂЃЈШчPearsonзлКЯеїЃЉЬсЙЉСЫбаОПЗЖЪНЃЛНвЪОаФСзжЌДњаЛдкдьбЊИЩЯИАћЮШЬЌЮЌГжжаЕФаТНЧЩЋЃЛНЈСЂЕФTazD75HаЁЪѓФЃаЭНЋГЩЮЊЮДРДвЉЮяЩИбЁЕФживЊЦНЬЈЁЃе§ШчТлЮФЭЈбЖзїепSimon J. ConwayЧПЕїЕФЃК"етВЛЪЧМђЕЅЕФЛљвђШБЪЇФЃаЭЃЌЖјЪЧецЪЕФЃФтЛМепЭЛБфЕААзЙІФмЕФЛюЬхбаОПЯЕЭГ"ЁЃЫцзХЖдTAZЗЧУИЙІФмЕФЩюШыЬНЫїЃЌBTHSЕФжЮСЦВпТдЛђНЋгРДШЋаТЭЛЦЦЁЃ
ЩњЮяЭЈЮЂаХЙЋжкКХ
жЊУћЦѓвЕеаЦИ
НёШеЖЏЬЌ | ШЫВХЪаГЁ | аТММЪѕзЈРИ | жаЙњПЦбЇШЫ | дЦеЙЬЈ | BioHot | дЦНВЬУжБВЅ | ЛсеЙжааФ | ЬиМлзЈРИ | ММЪѕПьбЖ | УтЗбЪдгУ
АцШЈЫљга ЩњЮяЭЈ
Copyright© eBiotrade.com, All Rights Reserved
СЊЯЕаХЯфЃК
дСICPБИ09063491КХ
