
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
scMKL:融合多组学数据的可解释性单细胞分析新方法揭示癌症生物学机制
【字体: 大 中 小 】 时间:2025年08月07日 来源:Communications Biology 5.1
编辑推荐:
研究人员针对单细胞多组学数据分析中预测能力与可解释性难以兼顾的挑战,开发了基于多核学习(scMKL)的创新算法。该方法整合scRNA-seq和scATAC-seq数据,通过路径诱导核和转录因子结合位点(TFBS)分组,在乳腺癌、前列腺癌等七大数据集中实现优于MLP/XGBoost的预测性能(AUROC 0.92-0.99),同时直接识别出ESR1/ER Early等关键调控通路,为癌症分型和治疗响应研究提供新见解。
在单细胞生物学蓬勃发展的今天,科学家们面临着一个关键矛盾:复杂的机器学习模型虽然预测精准,却如同"黑箱"难以解读;而简单的模型虽易于理解,又往往力不从心。这种困境在多组学数据整合分析中尤为突出――当需要同时解析转录组(scRNA-seq)和表观基因组(scATAC-seq)数据时,现有方法如MOFA+和Seurat/Signac要么依赖繁琐的数据预处理导致信息丢失,要么产生难以解释的潜在特征。更棘手的是,深度学习模型如自编码器虽然能捕捉非线性结构,但其决策过程缺乏透明度,甚至引发伦理担忧。
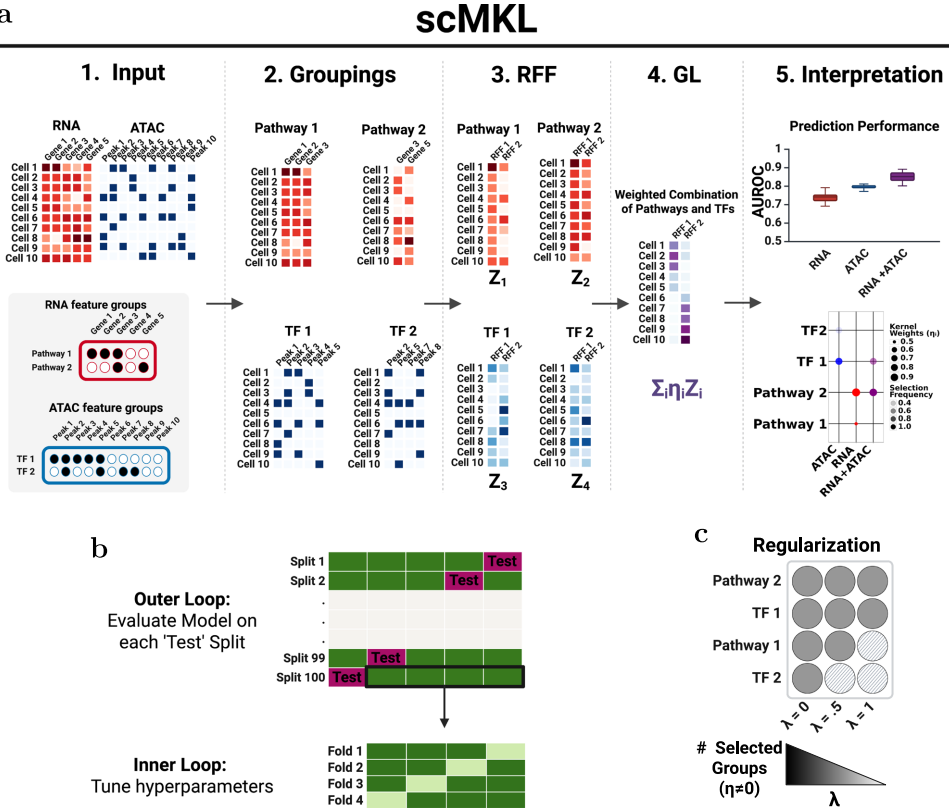
为破解这一难题,来自俄勒冈健康与科学大学(Oregon Health & Science University)的Cigdem Ak团队在《Communications Biology》发表了开创性研究。他们开发的scMKL算法巧妙融合多核学习(MKL)、随机傅里叶特征(RFF)和群组Lasso(GL),首次实现了单细胞多组学数据的可解释整合分析。这项研究不仅技术路线新颖,更在乳腺癌、前列腺癌等四种癌症类型中验证了其发现关键生物学通路的能力,为癌症机制研究和精准治疗提供了全新工具。
研究人员采用三大关键技术:首先构建基于Hallmark通路和JASPAR/Cistrome数据库的生物学先验知识分组;其次运用随机傅里叶特征将计算复杂度从O(N2)降至O(N);最后通过群组Lasso实现稀疏选择。实验数据涵盖7个数据集,包括MCF-7/T-47D乳腺癌细胞系(10x Multiome)、小淋巴细胞淋巴瘤(SLL)、前列腺癌(sciATAC-seq/scRNA-seq)和非小细胞肺癌(NSCLC)单细胞转录组,涉及6438-74,084个细胞。
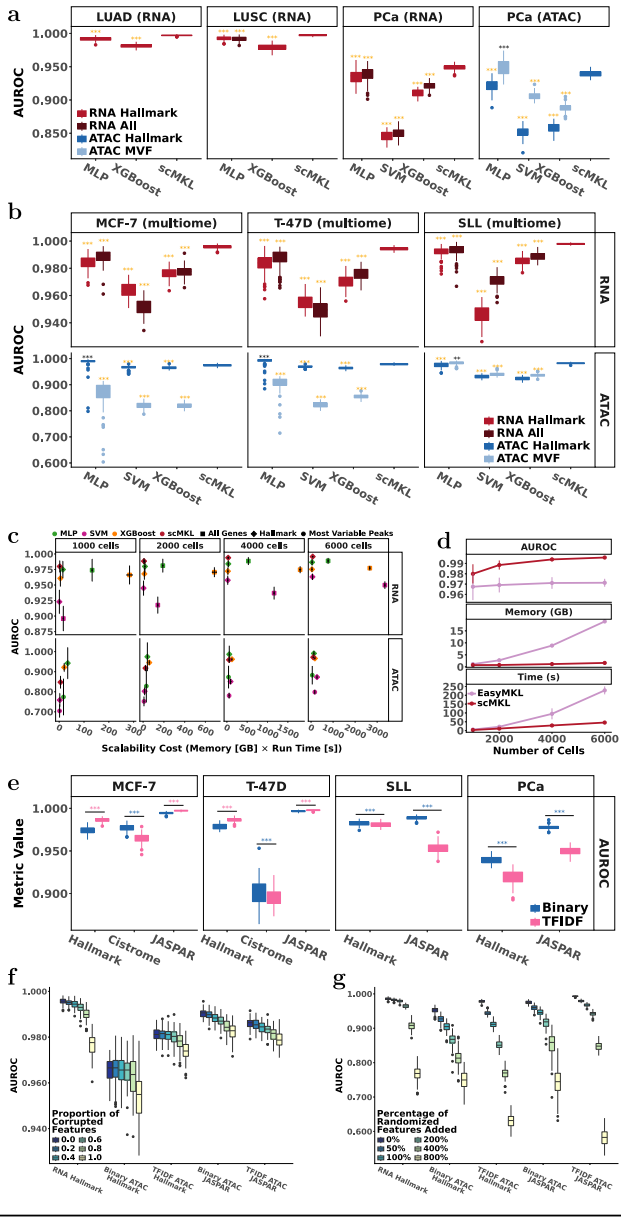
方法学突破方面,研究显示scMKL在保持可解释性的同时显著提升性能。与MLP、XGBoost和SVM相比,其AUROC提高5-15%,训练速度提升7倍,内存消耗减少12倍。特别在乳腺癌ER响应分析中,仅用Hallmark基因(4,384个)即超越使用全部基因(36,601个)的基准模型。通过系统扰动实验证实,基于生物分组的特征选择比传统"高变特征"方法更能捕捉真实信号。
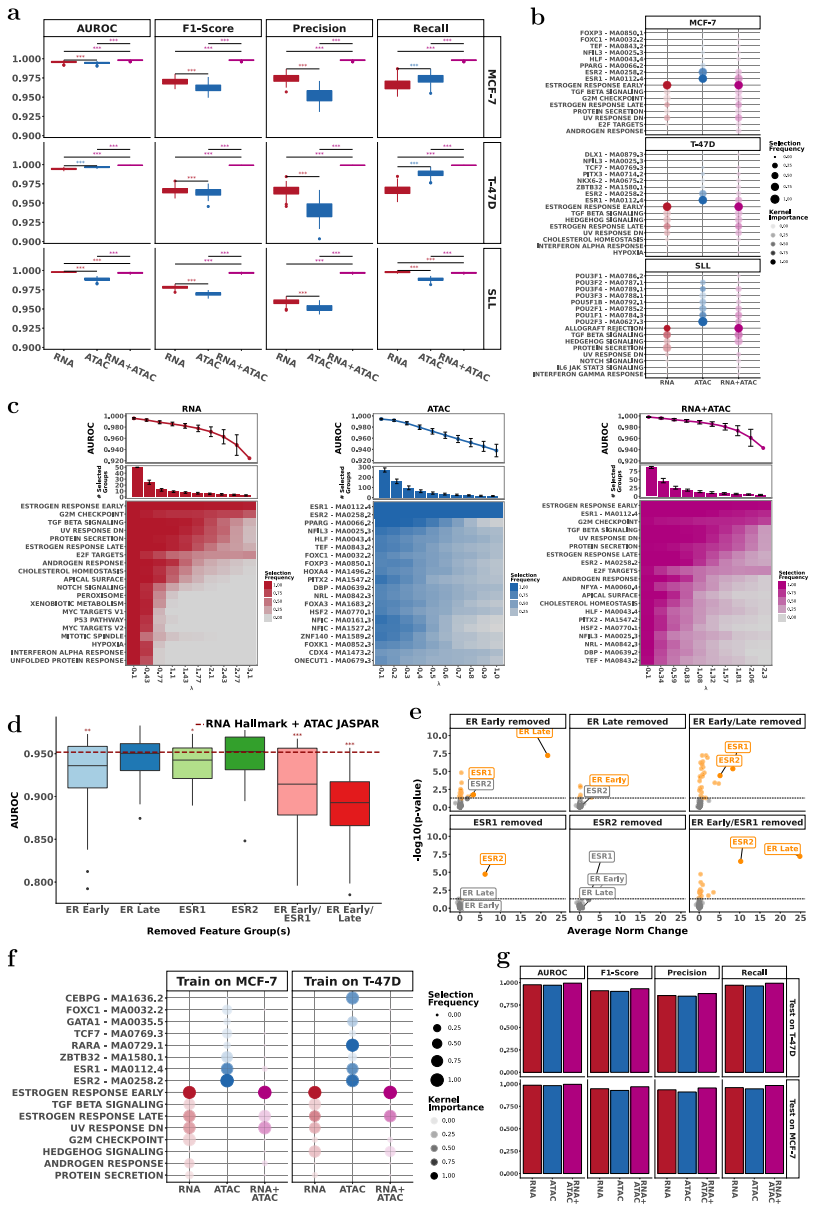
多组学整合部分揭示了跨模态协同效应。在MCF-7细胞中,RNA的ER Early通路与ATAC的ESR1结合位点被共同选择,AUROC达0.99。当人为剔除这些特征时,模型会自适应增强ESR2/ER Late等替代通路,印证了生物学合理性。转移学习实验证实,从MCF-7到T-47D细胞系的跨数据集预测保持0.96以上AUROC,且关键通路选择一致。
癌症生物学发现上,研究取得三项重要进展:(1)在前列腺癌sciATAC-seq数据中,scMKL区分低级别(Gleason 3)和高级别(Gleason 4)肿瘤,发现AR、WNT和Hedgehog通路权重差异,并鉴定出OTX2等神经内分泌分化相关转录因子;(2)在NSCLC分析中,ER Late通路特异性关联肺腺癌(LUAD),而MYC靶标富集于肺鳞癌(LUSC),与TCGA生存分析一致;(3)乳腺癌模型揭示MED12调控区域与ER Early通路基因存在显著基因组重叠,提示新型交叉调控机制。
这项研究的创新价值体现在三个维度:方法论上,首次将可解释性融入单细胞多组学整合分析;技术上,通过RFF和GL实现大规模计算;生物学上,发现ESR1-MED12等跨模态互作网络。其开源工具(scmkl 0.1.6)支持Hallmark/JASPAR等标准数据库,也可自定义特征组,为癌症异质性研究、生物标志物发现和耐药机制解析提供了灵活框架。未来拓展至DNA甲基化、蛋白质组等多组学整合,或将开启精准医学研究的新范式。
 生物通微信公众号
生物通微信公众号
知名企业招聘

