
-

生物通官微
陪你抓住生命科技
跳动的脉搏
综述:神经黑色素诱导的细胞应激与神经毒性在帕金森病发病机制中的作用
【字体: 大 中 小 】 时间:2025年08月09日 来源:Apoptosis 8.1
编辑推荐:
这篇综述系统阐述了神经黑色素(neuromelanin)在帕金森病(PD)中的双刃剑作用:既可通过螯合毒性物质发挥神经保护功能,又通过诱导氧化应激、线粒体功能障碍、自噬受损(autophagy)等机制加剧多巴胺能神经元退化。作者创新性提出靶向神经黑色素-毒性分子相互作用(如α-突触核蛋白(α-synuclein)聚集、铁死亡(ferroptosis))的潜在治疗策略,为PD干预提供新视角。
神经黑色素是一种主要存在于灵长类黑质(substantia nigra)和蓝斑(locus coeruleus)区域的深棕色色素,其形成涉及酪氨酸酶(tyrosinase)催化和多巴胺(dopamine)自氧化两条途径。值得注意的是,这种色素的年龄依赖性积累与帕金森病(PD)等神经退行性疾病密切相关。最新研究表明,神经黑色素可通过与铁离子、环境毒物等结合形成"特洛伊木马"式复合物,持续释放毒性成分导致神经元损伤。
多巴胺氧化过程中产生的中间产物多巴胺-o-醌(dopamine o-quinone)会迅速转化为氨基色素(aminochrome),后者通过单电子还原形成高反应性的白氨基色素-o-半醌自由基(leukoaminochrome o-semiquinone radical)。这一过程导致NADH耗竭并影响线粒体呼吸链ATP生成。
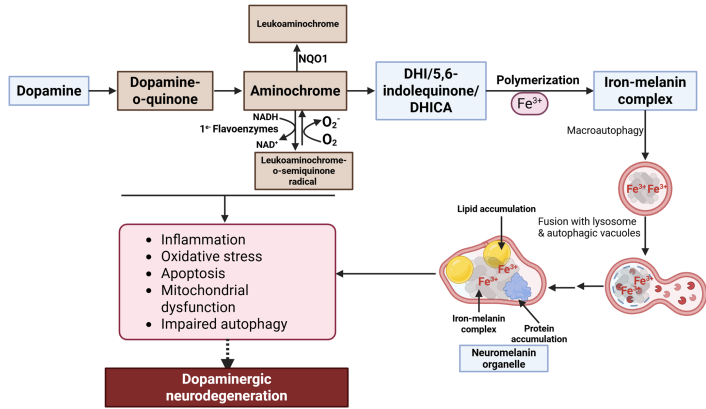
实验证据显示,氨基色素可诱导α-突触核蛋白原纤维形成,并通过抑制溶酶体功能使自噬流受阻。在分化的SH-SY5Y细胞中,氨基色素处理使ATP产量降低40%,同时显著增加铁转运蛋白DMT1表达,形成氧化应激恶性循环。
从死亡神经元释放的神经黑色素可通过激活小胶质细胞引发"炎症风暴"。动物实验证实,脑内注射神经黑色素可激活NF-κB和p38 MAPK通路,导致黑质区多巴胺摄取量下降65%。在分子层面,神经黑色素能破坏线粒体复合物I的超分子结构,诱发细胞色素c释放和caspase-3依赖性凋亡。
神经黑色素颗粒中检测到α-突触核蛋白共定位,两者相互作用形成正反馈环路:一方面α-突触核蛋白过表达可使黑质多巴胺水平升高30%,促进神经黑色素合成;另一方面神经黑色素积累会抑制泛素-蛋白酶体系统(UPS),导致α-突触核蛋白聚集体清除障碍。值得注意的是,携带A53T突变的α-突触核蛋白与神经黑色素共处理时,羟基自由基产量增加2.3倍。
MPP+与神经黑色素的结合常数高达108 M-1,这种强结合使毒性成分缓慢释放。类似地,烹饪产生的杂环胺(HAA)如harmane在神经黑色素存在时,可使SH-SY5Y细胞存活率降低58%。金属离子交互作用尤为关键,在PD患者黑质中,神经黑色素结合的铁离子饱和度达85%,显著高于健康对照。
铁螯合剂去铁胺(deferoxamine)可减少50%的神经黑色素合成,但其衍生物去铁酮(deferiprone)的临床试验结果矛盾:虽能保留脑组织体积,却因抑制酪氨酸羟化酶(TH)加剧运动症状。维生素E类似物Trolox通过阻断脂质过氧化链式反应,在动物模型中显示神经保护效应。
转录因子EB(TFEB)过表达可使细胞内神经黑色素密度降低40%,同时改善运动功能障碍。雷帕霉素(rapamycin)通过激活自噬显著减轻MPTP模型中的多巴胺能神经元丢失,其治疗阿尔茨海默病的临床试验正在进行中。
FKBP51抑制剂SAFit2在α-突触核蛋白转基因小鼠中可使 ubiquitin阳性包涵体减少70%。病毒载体介导的VMAT2过表达通过促进多巴胺囊泡封装,使氧化代谢产物降低3倍,这种"源头控制"策略可能比事后清除更有效。
神经黑色素作为PD病理过程中的关键分子节点,其与遗传因素(如LRRK2、PINK1)和环境因素的交互作用仍需深入解析。未来研究应聚焦于开发时空特异性调控手段,在保留其金属螯合功能的同时阻断毒性通路。基于生物标志物的早期干预和组合疗法(如自噬激活剂+铁死亡抑制剂)可能成为突破方向。




