
-

生物通官微
陪你抓住生命科技
跳动的脉搏
饮食诱导肥胖通过氧化应激和H3K4me3表观遗传修饰驱动造血干细胞髓系分化异常及炎症持续
【字体: 大 中 小 】 时间:2025年08月09日 来源:The Journal of Immunology 3.6
编辑推荐:
本研究揭示了高脂饮食(HFD)诱导肥胖通过氧化应激-H3K4me3-TLR4轴重编程造血干细胞(HSPCs)的表观遗传记忆,导致髓系分化异常和伤口修复障碍。研究人员采用CUT&Tag技术、流式细胞术和骨髓移植模型,发现肥胖小鼠HSPCs中KDM5A去甲基化酶活性降低导致H3K4me3在E2F靶点和Tlr4启动子区富集,环孢素A(CsA)干预可逆转这一过程。该研究为代谢性炎症的造血起源提供了新机制,发表于《The Journal of Immunology》。
在全球肥胖流行背景下,慢性低度炎症状态被认为是肥胖相关并发症的核心特征。传统观点认为这种炎症主要源于脂肪组织巨噬细胞的异常活化,但近年研究发现,肥胖甚至会改变造血干细胞的"出厂设置",使其持续产生促炎性免疫细胞。这种"造血记忆"现象背后的表观遗传机制,特别是组蛋白修饰如何介导代谢应激信号的长期保存,成为免疫代谢领域的重要科学问题。
美国纽约州立大学上州医科大学(State University of New York Upstate Medical University)的Kentaro Takahashi等研究人员通过高脂饮食(HFD)小鼠模型发现,肥胖小鼠的造血干细胞和祖细胞(HSPCs)移植给瘦小鼠后,竟能"传染"炎症表型――受体小鼠在肢体缺血后出现持续性炎症和肌肉修复障碍。这种跨代效应提示肥胖可能通过表观遗传重编程改变了HSPCs的固有属性。
研究采用多组学技术揭示了关键机制:
关键技术方法
骨髓移植和命运追踪实验(CD45异构体系统)
氧化应激检测(CellROX、MitoTracker)
表观遗传分析(CUT&Tag检测H3K4me3全基因组分布)
条件性基因敲除(KDM5A诱导型敲除小鼠)
体外干预实验(环孢素A、线粒体ROS清除剂处理)
主要研究结果
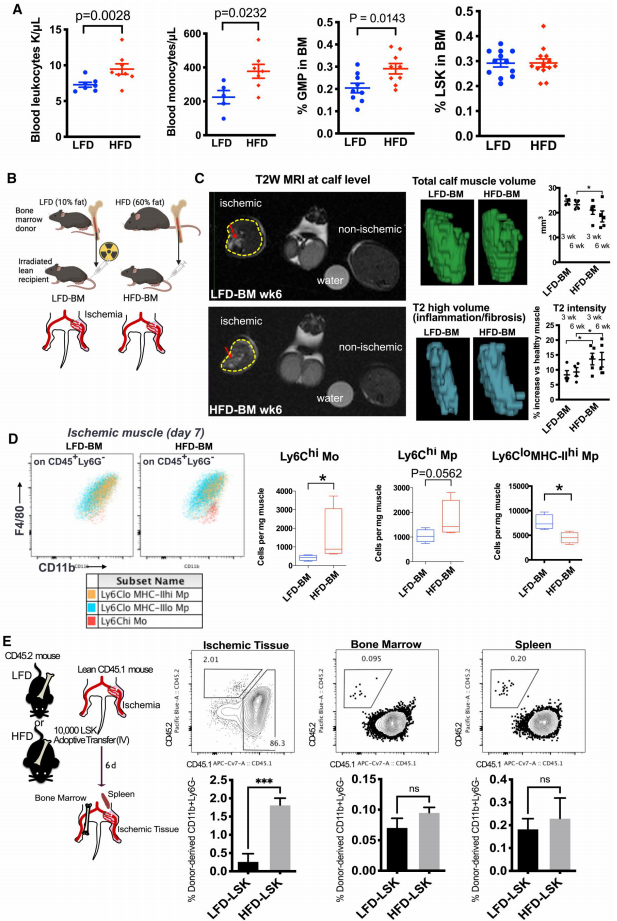
BM from HFD mice induces nonresolution in ischemic muscles
通过磁共振成像和流式分析发现,接受肥胖小鼠骨髓移植的瘦小鼠在肢体缺血后,肌肉炎症持续时间延长2周,促炎性Ly6Chi单核细胞增加40%,而修复型巨噬细胞减少。直接移植HFD-HSPCs后,受体肌肉中供体来源的炎症细胞增加3倍,证实肥胖通过HSPCs内在改变影响炎症结局。
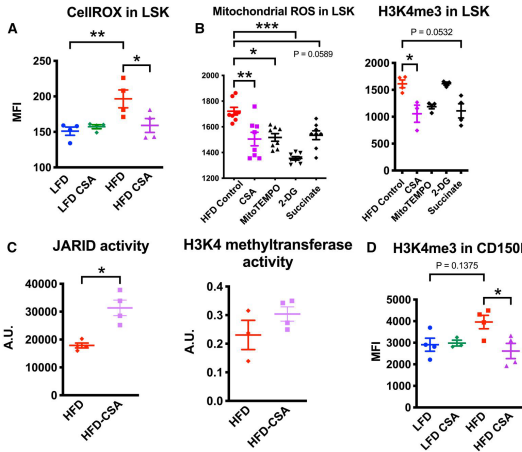
HFD increases oxidative stress and H3K4me3 in HSPCs
流式检测显示HFD-HSPCs线粒体ROS水平升高2.5倍,伴随H3K4me3修饰增加(而非H3K4me1或H3K27ac)。酶活检测揭示KDM5家族去甲基化酶活性降低60%,而甲基转移酶活性不变,提示氧化应激通过抑制去甲基化导致H3K4me3累积。
CsA reverses HFD-induced HSPC signatures
环孢素A处理使HFD-HSPCs的ROS和H3K4me3水平恢复正常,特异性恢复KDM5去甲基化酶活性。CD150+LSK(最原始造血干细胞)中H3K4me3降低35%,证实干预靶向干细胞区室。
CUT&Tag reveals H3K4me3 dynamics during myelopoiesis
表观基因组分析发现:
LSK→GMP分化时E2F靶点(如Ccnb2、Cdc6)的H3K4me3富集增强
髓系特征基因(Mpo、Cebpa等)伴随染色质开放状态出现H3K4me3上调
HFD-HSPCs中Tlr4启动子区H3K4me3信号增强2倍
KDM5A knockout mimics HFD phenotype
诱导性敲除KDM5A后:
HSPCs中H3K4me3水平升高
TLR2/4配体刺激的髓系分化增强3倍
皮肤损伤后脾脏单核细胞增加50%
证实KDM5A失活是肥胖表型的核心环节
研究结论与意义
该研究建立了"代谢应激-表观遗传-造血输出"的完整调控轴:高脂饮食→HSPCs氧化应激→KDM5A活性抑制→H3K4me3在Tlr4/E2F靶点累积→TLR4高反应性和髓系分化偏倚→组织修复障碍。创新性发现包括:
首次证实H3K4me3在肥胖相关造血记忆中的枢纽作用
揭示KDM5A去甲基化酶是代谢-表观遗传交叉对话的关键节点
提供环孢素A干预表观遗传重编程的治疗新思路
该研究为理解慢性炎症的造血起源提供了新范式,提示靶向HSPCs表观遗传调控可能是治疗肥胖相关并发症的新策略。未来研究需探索组织特异性炎症中HSPCs的记忆特征,以及如何精确调控组蛋白修饰而不影响正常造血功能。




