
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
基于混合矩特征建模的piRNA集成识别策略:TranspoPred算法在转座子衍生piRNA预测中的突破性应用
【字体: 大 中 小 】 时间:2025年08月19日 来源:Scientific Reports 3.9
编辑推荐:
本研究针对转座子衍生piRNA(piwi-interacting RNAs)预测精度不足的问题,开发了集成混合矩特征建模的TranspoPred算法。通过整合位置相关、频率和统计矩特征,结合Bagging/Boosting/Stacking三种集成学习方法,在人类、小鼠和果蝇数据集上实现了99%的预测准确率(ACC)和Matthew相关系数(MCC),显著优于现有工具piRNApredictor和Piano。该成果为研究piRNA在转座子沉默和基因组稳定性维持中的调控机制提供了高效计算工具。
在生命科学的微观世界里,有一类长度仅26-32个核苷酸的小RNA分子――piRNA(piwi-interacting RNAs),它们如同基因组的"守护者",专门沉默转座子(transposable elements)的活性,维持生殖细胞基因组的稳定性。然而这些"分子卫士"的识别却面临巨大挑战:传统湿实验方法存在低通量、高成本的局限,而现有计算工具如piRNApredictor和Piano的预测准确率普遍低于85%。更棘手的是,piRNA序列具有高度异质性,缺乏保守的二级结构特征,使得计算机建模难度倍增。
针对这一难题,来自巴基斯坦管理技术大学(University of Management and Technology)的Mansoor Ahmed Rasheed团队在《Scientific Reports》发表了创新性研究成果。研究人员开发了名为TranspoPred的集成预测系统,通过融合统计矩特征与深度学习网络,首次在人类、小鼠和果蝇三个物种数据集上实现了近乎完美的预测性能。这项突破不仅为piRNA研究提供了强大工具,更揭示了转座子衍生piRNA的序列特征规律。
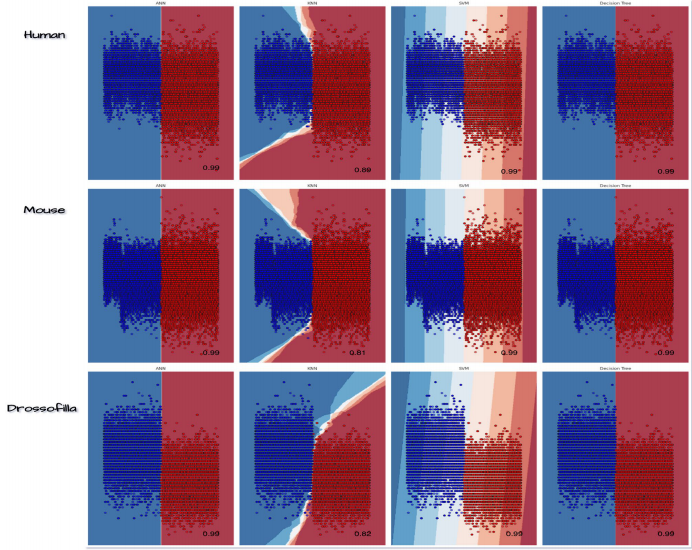
研究团队采用了多模态特征提取策略:首先通过位置相对关联矩阵(PRIM)和反向位置相对关联矩阵(RPRIM)捕获核苷酸的空间分布特征;接着计算原始矩、中心矩和Hahn矩等统计特征,将41bp的RNA序列转化为522维特征向量。在模型构建阶段,创新性地并行应用了三种集成学习方法:Bagging(随机森林/极端随机树)、Boosting(XGBoost/AdaBoost)和Stacking(以神经网络为元学习器)。特别值得注意的是,团队建立了严格的评估体系,采用2×5折交叉验证、10折交叉验证和独立测试集三重验证,确保结果的可靠性。

研究结果展现出令人振奋的性能表现。在人类数据集上,Stacking集成模型在独立测试中取得ACC=0.99、MCC=0.99的完美成绩,敏感性(Sn)和特异性(Sp)均达99%。值得注意的是,该模型对果蝇数据的预测同样出色(AUC=0.99),且在小鼠数据集上保持98%的准确率,证明其跨物种适用性。通过决策边界可视化分析发现,TranspoPred能清晰区分piRNA与非piRNA的序列特征空间,这解释了其超高精度的内在机制。
与现有工具的直接对比凸显了TranspoPred的优越性:在人类数据测试中,其AUC值(0.99)显著高于piRNApredictor(0.898)和Piano(0.596)。更引人注目的是,模型在保持高精度的同时展现出强大的泛化能力――当使用人类数据训练的模型直接预测小鼠piRNA时,仍获得91%的准确率,暗示不同物种piRNA可能存在保守特征模式。
这项研究在方法学和应用层面均具有重要价值。在技术上,首创的混合矩特征建模框架为其他非编码RNA识别提供了新思路;在生物学意义上,精确的piRNA预测工具将加速转座子调控网络解析,为不孕症、癌症等生殖细胞相关疾病研究提供新视角。研究人员已将全部代码和数据集开源,这种开放科学的态度将进一步推动piRNA研究社区的发展。未来工作可探索更多物种的piRNA特征规律,并将该框架扩展到piRNA-靶基因互作预测等领域。
本研究也存在若干值得探讨的局限:特征工程过程计算复杂度较高;模型对长度异常(<24nt或>32nt)的piRNA变体识别效果有待验证。此外,虽然团队通过排除tRNA/snoRNA等干扰因素提高了特异性,但真实生物样本中piRNA与其他小RNA的分子互作可能带来新的挑战。这些问题的解决将推动下一代piRNA预测工具的发展。
生物通微信公众号
知名企业招聘

