
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
淋巴毒素驱动的小鼠肌炎与自噬障碍的相互强化机制及其在包涵体肌炎中的病理意义
【字体: 大 中 小 】 时间:2025年08月26日 来源:Brain 11.7
编辑推荐:
本研究针对包涵体肌炎(IBM)中炎症与蛋白稳态失衡的相互作用机制,通过构建骨骼肌特异性表达淋巴毒素(LTα/β)的转基因小鼠模型,首次揭示了LT信号通路通过激活NF-κB引发慢性肌炎,并导致自噬溶酶体通路先激活后耗竭的双向调控模式。研究发现遗传性自噬障碍(Atg5敲除)会加剧LT诱导的肌炎表型,形成类似人类IBM特征的管丝状包涵体,为理解炎症与退行性病变的恶性循环提供了新视角。该成果为开发靶向LT信号和自噬通路的联合治疗策略奠定了理论基础。
在神经肌肉疾病领域,包涵体肌炎(IBM)始终是个令人困惑的谜题。这种主要影响中老年人的进行性肌肉疾病,同时表现出炎症和退行性双重特征――就像一场在肌肉组织中同时上演的"免疫风暴"和"蛋白质灾难"。患者肌肉中既有CD8+T细胞的浸润,又堆积着β-淀粉样蛋白、磷酸化tau等典型神经退行性疾病相关的蛋白聚集体。更棘手的是,常规免疫治疗对IBM几乎无效,这暗示着其发病机制可能比传统自身免疫性疾病更为复杂。
科学家们长期争论:究竟是炎症引发了蛋白代谢紊乱,还是蛋白稳态失衡触发了炎症?为破解这个"鸡生蛋还是蛋生鸡"的难题,Juliane Bremer等研究者独辟蹊径,将目光聚焦于淋巴毒素(LT)信号通路。这个选择绝非偶然――既往研究发现LTα/β在人类炎性肌病中异常激活,且能通过NF-κB通路同时调控炎症和细胞应激反应。但LT如何具体参与IBM发生发展?它与自噬障碍如何相互作用?这些关键问题一直悬而未决。
研究团队首先通过分析人类肌炎样本,确认了LT信号通路相关基因的显著上调。在此基础上,他们创新性地构建了骨骼肌特异性共表达LTα和LTβ的转基因小鼠(HSA-LTα/β),并联合肌肉特异性Atg5敲除技术,通过多组学分析、高分辨率显微成像和行为学测试等手段,系统揭示了LT驱动肌炎与自噬障碍相互强化的分子机制。
关键技术包括:(1)建立HSA启动子驱动的LTα/β双转基因小鼠模型;(2)通过Ckmm-Cre与Atg5flox/flox杂交获得肌肉特异性自噬缺陷小鼠;(3)采用RNA-seq和qPCR分析转录组变化;(4)电子显微镜观察超微结构特征;(5)线粒体DNA长读长测序技术;(6)肌肉MRI体积定量分析;(7)自动化行为学监测系统评估运动功能。人类样本来源于武装部队病理学研究所(AFIP)的存档组织。
【淋巴毒素和靶基因在人类IIM中的表达】研究团队通过qPCR检测发现,多发性肌炎(PM)中LTα显著上调,而皮肌炎(DM)和IBM中LT靶基因CCL19、CXCL10等明显升高。原位杂交证实LTα、LTβ及其受体在IBM患者肌纤维和炎症浸润细胞中均有表达,为后续动物模型构建提供了人类疾病依据。
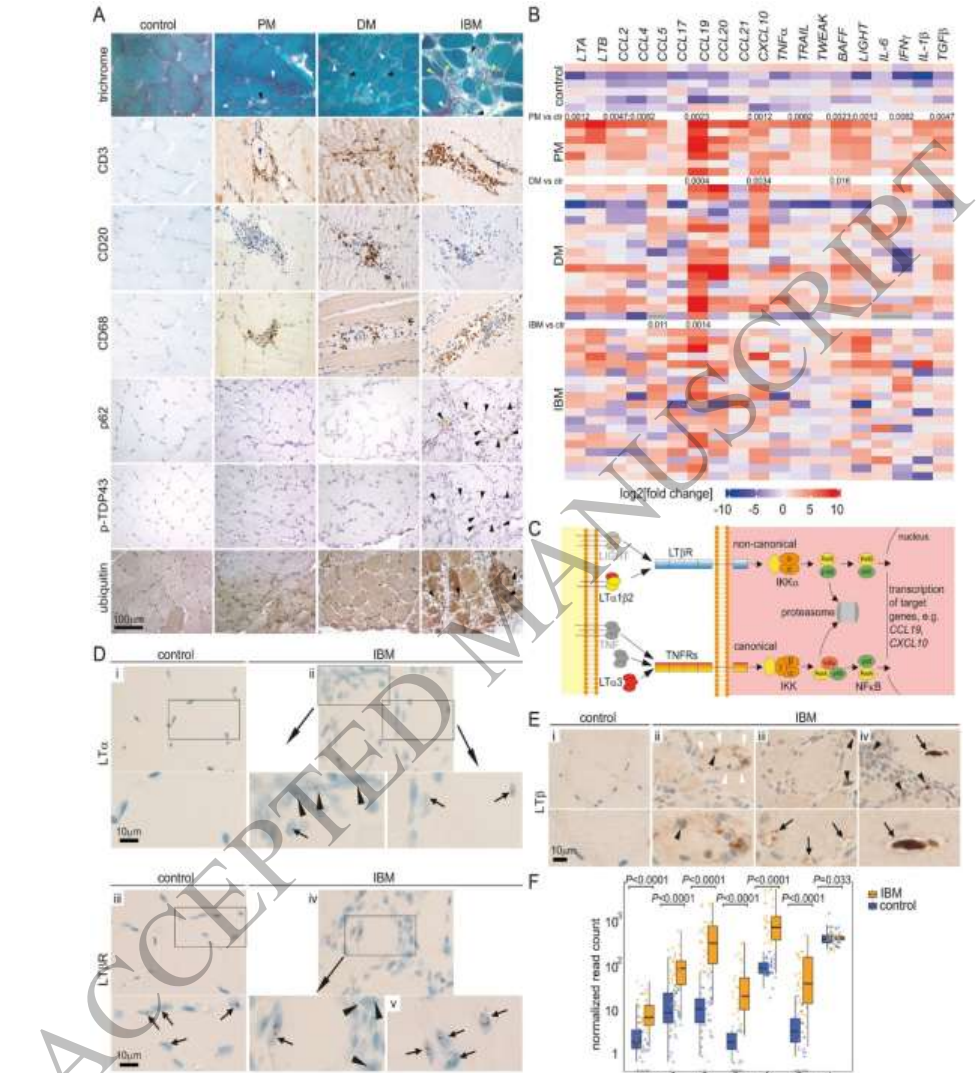
【HSA-LTα/β转基因小鼠的表型特征】转基因小鼠表现出与人类肌炎相似的病理改变:肌纤维大小变异增大、进行性肌萎缩和运动功能减退。免疫组化显示肌内膜浸润以B220+B细胞和CD68+巨噬细胞为主,伴随MHC I上调和内质网应激标志物表达增加。有趣的是,自噬相关基因Atg5和Becn1在3-6月龄时上调,而在10月龄时转为下调,提示自噬功能呈现先激活后耗竭的动态变化。
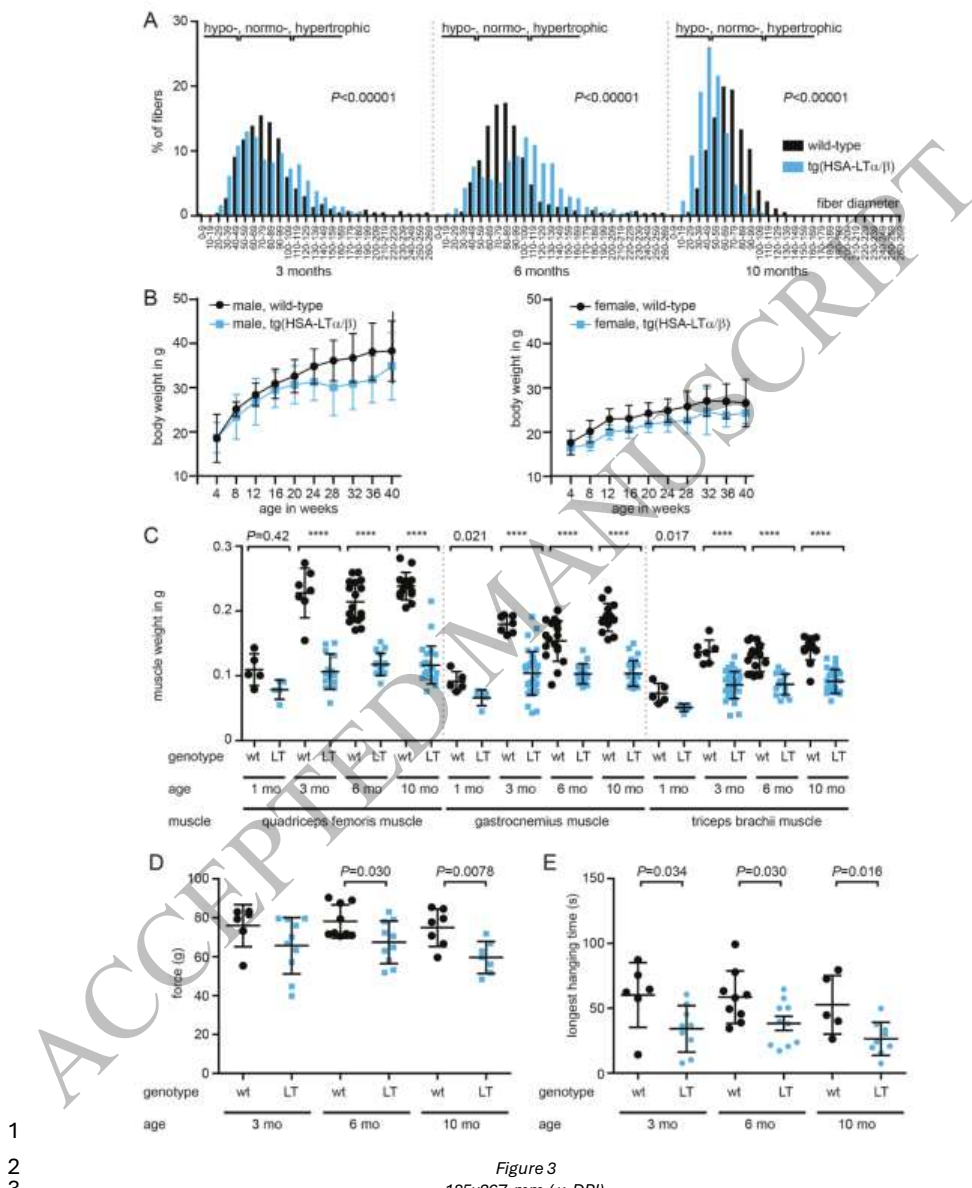
【慢性肌炎诱导细胞器应激和线粒体异常】RNA-seq分析显示,转基因小鼠肌肉中内质网应激相关基因(如Chop、Atf3)和抗氧化酶(Gpx1、Sod3)显著上调,但线粒体相关基因随年龄增长未能正常上调。电镜观察到线粒体肿胀等轻微异常,但未见典型类晶体包涵体。这些发现提示LT引发的慢性炎症可能导致线粒体功能代偿不足。
【自噬缺陷加重LT诱导的肌炎表型】当在HSA-LTα/β小鼠中引入Atg5敲除(HSA-LT;CreAtg5)后,表型发生质的改变:出现大量泛素化、p62阳性的蛋白聚集体,电镜下可见直径约17nm的管丝状包涵体――这与人类IBM特征性病理改变高度相似。特别值得注意的是,COX-SDH双染色首次在该模型中发现线粒体酶活性镶嵌现象,提示自噬障碍加剧了线粒体功能异常。
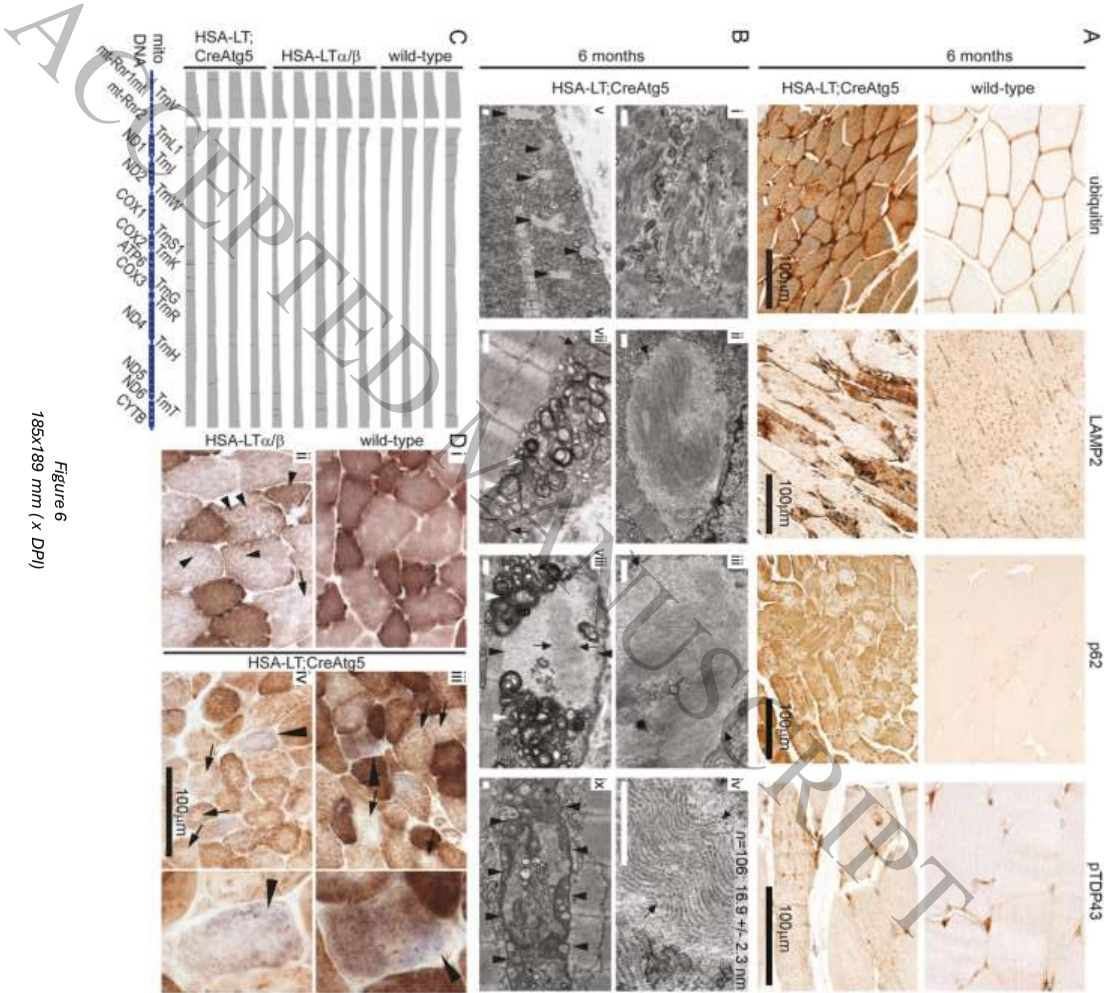
【治疗抵抗性的分子基础】尝试用泼尼松和抗Thy1.2抗体治疗转基因小鼠,均未能改善运动功能。分子分析发现,即使T细胞被清除,肌纤维内蛋白聚集和退行性改变依然进展。这可能与肌肉干细胞(Pax7+卫星细胞)减少导致的再生缺陷有关,也与高度分化的KLRG1+细胞毒性T细胞的持续存在相关。
这项研究的重要发现在于:首次在动物模型中重现了IBM的两大核心特征――慢性淋巴细胞性肌炎和蛋白聚集体形成,并阐明其通过LT-NF-κB信号轴与自噬障碍形成的恶性循环机制。特别具有临床意义的是,研究发现单纯抑制炎症不能逆转已经建立的退行性病变,这为解释IBM对免疫治疗不敏感提供了实验依据。研究还提出Tid1(肿瘤性想象盘1)下调可能是连接线粒体功能障碍、β-淀粉样蛋白产生和炎症放大的关键分子节点。
该成果为理解炎症性肌病的发病机制提供了全新视角:LT信号通路激活不仅是炎症的驱动因素,更是蛋白稳态失衡的始作俑者;而自噬功能障碍也不仅是退行性变的后果,还能通过上调CCL5等趋化因子反馈强化炎症微环境。这种双向互作机制的揭示,将推动针对LT信号和自噬通路的联合治疗策略开发,为目前缺乏有效治疗的IBM患者带来新希望。
生物通微信公众号
知名企业招聘

