
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
综述:阿尔茨海默病中自噬与凋亡的交互作用:分子机制与治疗靶点
【字体: 大 中 小 】 时间:2025年08月27日 来源:Discover Neuroscience
编辑推荐:
这篇综述深入探讨了自噬(autophagy)与凋亡(apoptosis)在阿尔茨海默病(AD)中的复杂调控网络,系统梳理了PI3K/Akt/mTOR、AMPK、p38 MAPK等关键信号通路对Aβ和tau病理的双向调控作用,为开发基于自噬-凋亡交互靶点的疾病修饰疗法提供了理论框架。
自噬与AD病理的关联
自噬-溶酶体途径(ALP)作为细胞内主要降解系统,通过清除错误折叠蛋白和受损细胞器维持神经元稳态。在AD早期,自噬障碍导致β-淀粉样蛋白(Aβ1-42)和过度磷酸化tau蛋白积累;晚期则因溶酶体功能障碍出现自噬体堆积。转录因子EB(TFEB)作为调控核心,其核转位可促进Aβ清除,但持续内质网应激(ER stress)引发的过度自噬反而加速神经元死亡。铁代谢异常通过NCOA4介导的铁蛋白自噬(ferritinophagy)诱发铁死亡,进一步加剧神经退行性变。
凋亡在AD中的驱动作用
p38 MAPK和JNK通路是Aβ诱导神经元凋亡的关键媒介,通过磷酸化p53、激活Bax等促凋亡蛋白触发 caspase-3级联反应。线粒体靶向的Aβ积累破坏电子传递链,促使细胞色素c释放并打开线粒体通透性转换孔(mPTP),而亲环蛋白D(Cyclophilin D)缺失可显著减轻这种损伤。值得注意的是,MAPK通路呈现双面性:ERK增强突触可塑性,而p38/JNK主导凋亡信号传导。
核心信号通路的调控网络
PI3K/Akt/mTOR轴通过抑制GSK-3β减少tau磷酸化和BACE-1表达,但过度激活会阻碍ULK1介导的自噬起始。AMPK作为能量传感器,通过磷酸化ULK1Ser317和抑制mTORC1促进自噬体形成,其激活剂如细叶远志皂苷可降低Aβ25-35的神经毒性。SIRT1通过去乙酰化p53和FOXO抑制凋亡,同时调控NF-κB减轻神经炎症。PINK1/Parkin通路缺陷导致线粒体自噬障碍,加速Aβ聚集和能量危机。
治疗策略与未来方向
靶向NLRP3/caspase-1/IL-1β轴既可抑制神经炎症,又能恢复自噬流量。电针抑制p38 MAPK可减少tau磷酸化,而白藜芦醇通过激活SIRT1发挥神经保护作用。时空特异性调控成为关键――早期增强自噬清除Aβ,晚期则需抑制过度自噬。整合血浆生物标志物和神经影像技术将有助于监测靶向治疗响应。
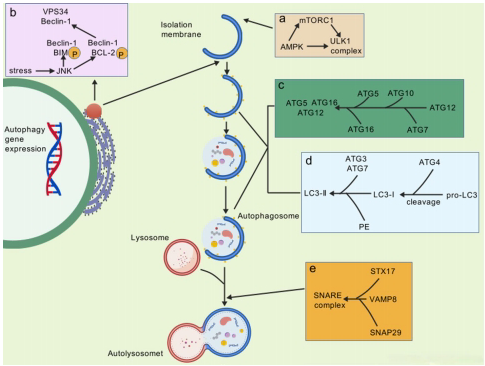
该示意图生动展示了自噬过程的分子机制:AMPK-mTOR轴调控ULK1复合体形成,Beclin-1/VPS34生成PI3P促进囊泡扩张,ATG12-ATG5-ATG16L复合物完成聚合,LC3-II插入自噬体膜,最终通过STX17/SNAP29/VAMP8介导的膜融合形成自溶酶体。这种精细的级联反应为开发多靶点干预策略提供了可视化依据。
生物通微信公众号
知名企业招聘

