
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
scSorterDL:基于深度神经网络集成pLDA模型的单细胞RNA测序数据分类新方法
【字体: 大 中 小 】 时间:2025年09月02日 来源:Briefings in Bioinformatics 7.7
编辑推荐:
本研究针对单细胞RNA测序(scRNA-seq)数据高维度、高稀疏性导致的细胞类型注释难题,开发了集成惩罚线性判别分析(pLDA)、群体学习(swarm learning)和深度神经网络(DNN)的scSorterDL算法。通过生成300个随机数据子集训练pLDA模型,并利用DNN整合模型输出,在33个跨平台实验中准确率达0.98(交叉验证)和0.91(跨平台),显著优于9种现有工具。该研究为单细胞数据分析提供了高效可靠的自动化注释方案,相关代码已开源。
单细胞RNA测序(scRNA-seq)技术的突破性发展,让科学家们得以窥见组织内单个细胞的基因表达图谱,这为理解细胞异质性打开了新窗口。然而,这项技术也带来了甜蜜的烦恼――如何准确给海量单细胞数据贴上类型标签?传统的细胞注释方法在面对高维度(通常含2-3万个基因)和极端稀疏(平均每个基因在70-90%细胞中零表达)的单细胞数据时,就像用渔网捞针,既容易漏掉关键信息,又难以避免技术噪音的干扰。更棘手的是,不同实验平台产生的数据存在显著差异,这使跨数据集比较变得异常困难。
针对这些挑战,来自加拿大维多利亚大学等机构的研究团队在《Briefings in Bioinformatics》发表了创新性解决方案。他们开发的scSorterDL系统,巧妙融合了三种核心技术:首先采用惩罚线性判别分析(pLDA)处理高维数据,通过协方差矩阵正则化解决"维度灾难";其次运用群体学习策略,从随机采样的80%细胞和√p+70基因(p为总基因数)的子集中训练300个pLDA模型;最后构建深度神经网络(DNN)架构,通过定制化损失函数整合各模型输出。研究特别采用GPU并行计算加速处理,使算法在保持精度的同时具备处理百万级细胞的能力。
预处理流程
系统通过Wilcoxon秩和检验筛选每种细胞类型前400个差异基因,采用log-normalization标准化(缩放因子10,000)处理数据。测试显示基因筛选数量主要影响计算速度,对精度无显著影响。
群体LDA模块性能
在胰腺数据集测试中,基因子集平均重叠率仅2.01%,而细胞重叠率达66.7%,证明该方法能有效捕获数据多样性。比较四种采样策略发现,均匀采样在保持模型多样性方面表现最优。
惩罚LDA设计
通过引入收缩估计量Σ(β)=(1-β)Σ+βTr(Σ)I/p,解决基因数远超样本数导致的矩阵奇异问题。判别函数δk(x)=xTΣ(β)-1μk-1/2μkTΣ(β)-1μk+log(πk)将数据投影到K-1维空间,兼顾效率与可解释性。
集成模块创新
突破性地采用双阶段DNN架构:先用独立DNN处理各pLDA模型的判别分数,生成初步预测概率pk(m);再通过细胞类型特异性加权投票层(权重wmk由softmax学习)整合结果。定制化损失函数L(x,y)=αC(F(g(1)(δ(x)),...,g(M)(δ(x))),y)+(1-α)ΣC(g(m)(δ(x)),y)通过α参数平衡最终输出与中间结果监督,类似残差网络的跳跃连接机制。
在33个基准实验中,scSorterDL展现出卓越性能。交叉验证实验使用13个数据集(涵盖人/鼠的胰腺、大脑、PBMC等组织),平均准确率达0.98,较第二名Seurat提升13%。
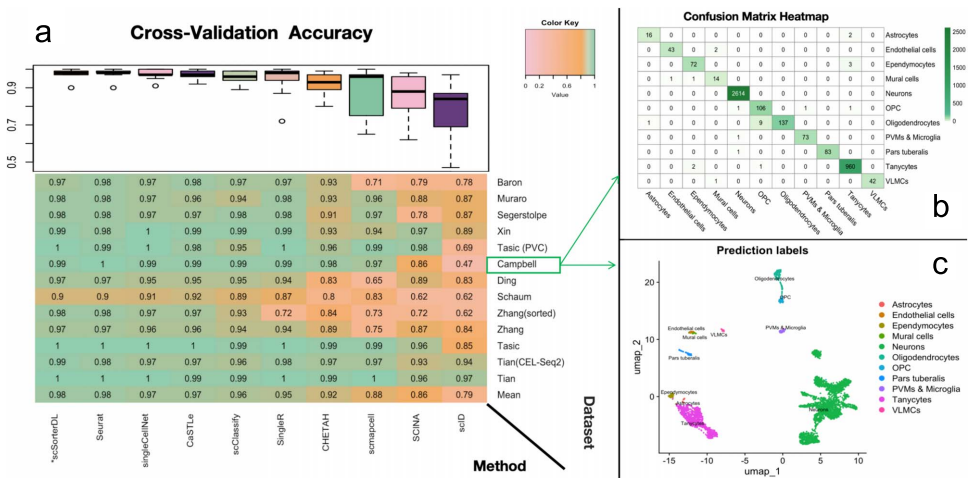
跨平台测试更具挑战性,涉及20对采用不同协议(如10X Chromium与SMART-Seq2)生成的数据集。scSorterDL以0.89的平均准确率领先,在PBMC数据(inDrop→10X转换)中准确识别出相邻UMAP簇中的相似细胞类型。
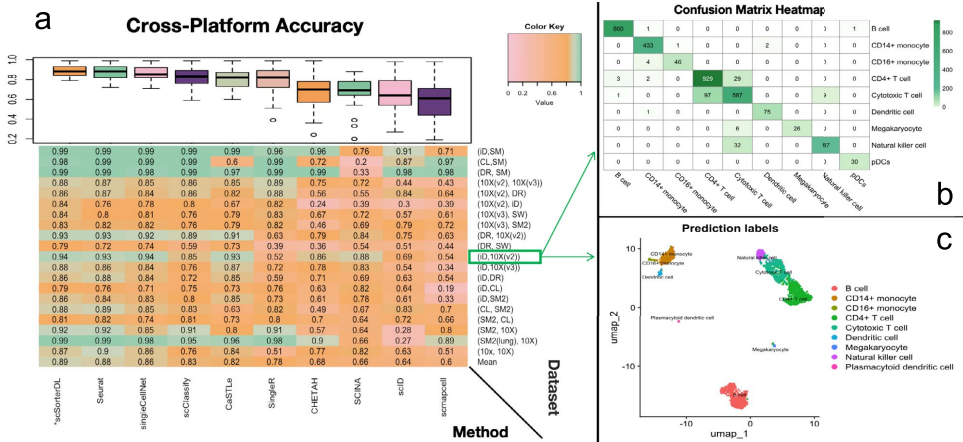
研究同时揭示了技术局限性:当前版本依赖预处理基因筛选,未来计划整合自编码器实现端到端训练;在6个癌症数据集测试中(平均精度0.88),虽然表现优于多数基线方法,但对肿瘤异质性的处理仍有提升空间。作者建议每个细胞类型至少需要20-30个参考细胞以保证统计稳定性,这与Soneson等关于单细胞分析样本量的研究结论一致。
这项研究的突破性价值体现在三个方面:方法论上,首次将群体学习与深度学习有机结合,为高维生物数据建模提供新范式;技术上,通过GPU加速实现大规模单细胞数据的实时分析;应用层面,其开源工具(GitHub可获取)可无缝整合到现有单细胞分析流程。正如作者强调的,该框架可扩展至其他机器学习场景,特别是需要处理特征和样本双重采样的复杂问题。随着单细胞图谱项目的全球推进,这种兼具准确性、鲁棒性和可扩展性的注释方法,将为解密细胞宇宙提供更精准的"罗盘"。
生物通微信公众号
知名企业招聘

