
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
基于KMA邻近评分的纳米孔与Illumina数据无组装分型技术突破
【字体: 大 中 小 】 时间:2025年09月02日 来源:NAR Genomics and Bioinformatics 2.8
编辑推荐:
本研究针对第三代测序技术(ONT)在高度相似参考序列分型中的多映射问题,开发了KMA v1.4.0的邻近评分算法。通过MLST测试验证,该技术使ONT数据分型准确率达99.8%,计算资源仅需智能手机级别,为资源有限实验室提供了高效解决方案。
微生物分型技术正经历从传统分子方法到高通量测序的革命性转变。牛津纳米孔技术(Oxford Nanopore Technologies, ONT)凭借其长读长优势,使测序设备能够进入普通实验室,但计算基础设施仍是主要障碍。尤其在进行多基因座序列分型(Multilocus Sequence Typing, MLST)时,高度相似的等位基因常因测序错误导致多映射问题,传统ConClave算法对第三代测序数据的处理效果欠佳。
针对这一技术瓶颈,Philip T.L.C. Clausen团队开发了KMA(k-mer alignment)算法的升级版(v1.4.0)。该技术通过创新的邻近评分(proximity scoring)机制,在保持智能手机级计算效率的同时,成功解决了ONT数据在冗余数据库中的精确分型难题。这项突破性研究已发表在《NAR Genomics and Bioinformatics》期刊。
研究采用k-mer签名哈希映射和双步比对策略,首先通过14-mer最小化索引筛选候选参考序列,再通过种子-链-比对程序验证。核心创新是改进的ConClave算法,引入ε参数(0-1]动态调整候选参考范围,并利用共识序列进行二次分配。评估使用137株覆盖14个菌种的细菌分离株,包括不同ONT流动槽版本(r9.4.1/r10.3/r10.4.1)和Illumina对比数据。
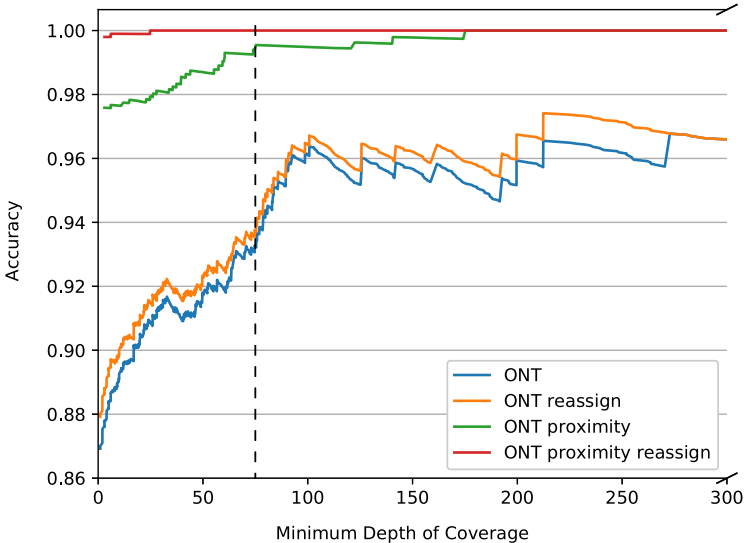
"MLST of long and short sequence data"部分显示,启用邻近评分后ONT数据分型准确率从87.93%提升至99.80%。典型案例如大肠杆菌purA_7等位基因的误分型,通过共识序列重新比对得到纠正。特别值得注意的是,团队发现金黄色葡萄球菌MLST数据库中arcC_755等位基因可能源于454测序的同聚物错误,经与数据库管理者确认后修正。
"Computational requirements"部分证实,该技术在2015款MacBook Pro(16GB内存)上处理ONT数据仅需39秒/样本,峰值内存106MB,效率显著优于Winnowmap2和Krocus等工具。
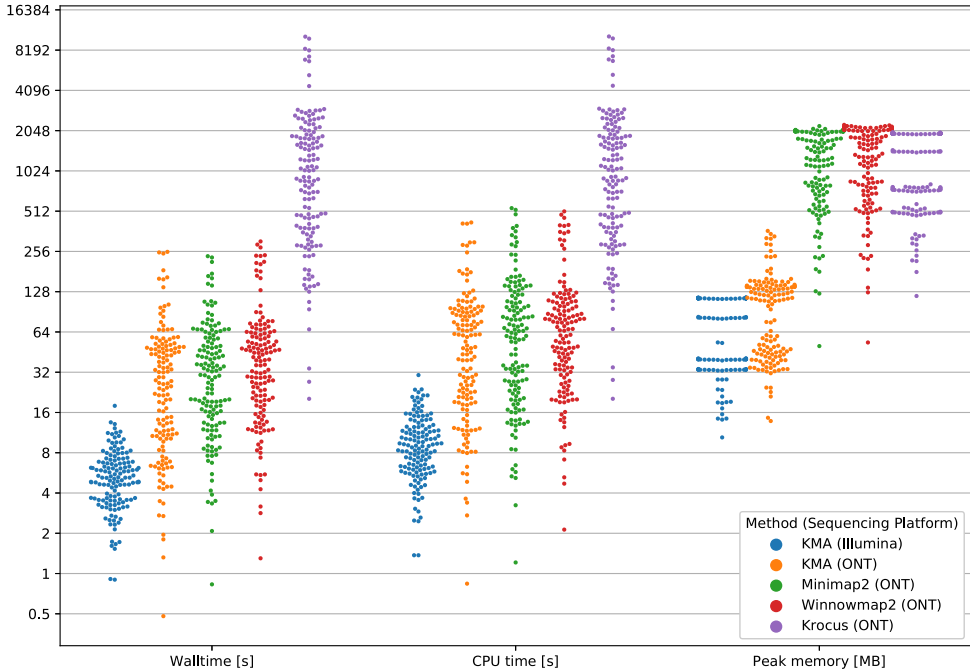
讨论部分强调,r10.4.1流动槽在保持高产量的同时提高了准确性,使分型深度要求降低。研究还发现约2%的低丰度匹配可能源于ONT索引跳跃现象。该方法成功克服了Winnowmap2等工具在参考序列间多映射读长分辨上的局限性,同时避免了Krocus工具在新菌种识别上的不确定性。
这项研究的技术突破具有双重意义:一方面使资源有限实验室能够进行高质量微生物分型,促进全球微生物监测网络的扩展;另一方面为抗菌素耐药基因检测等复杂性状分析奠定了基础。研究过程中发现的数据库错误也凸显了该技术在数据库质量控制方面的附加价值。随着ONT技术持续发展,这种高效分型方法将在传染病防控、医院感染监测等领域发挥更大作用。
生物通微信公众号
知名企业招聘

