
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
DRAGoN:基于Nextflow的高效DRUG-seq数据分析流程及其在药物基因组学中的应用
【字体: 大 中 小 】 时间:2025年09月09日 来源:Bioinformatics Advances 2.8
编辑推荐:
针对现有DRUG-seq(Digital RNA with perturbation of Genes)分析流程灵活性不足、计算资源消耗大的问题,Merck团队开发了基于Nextflow的DRAGoN流程。该流程通过前期解复用(demultiplexing)实现并行化处理,显著提升分析效率,在保持准确性的同时将运行时间缩短至竞争方法的1/3,为高通量药物筛选提供可靠工具。研究发表于《Bioinformatics Advances》。
在药物研发和基础研究中,高通量转录组技术DRUG-seq(Digital RNA with perturbation of Genes)正成为揭示小分子化合物和基因扰动效应的利器。然而,随着测序通量的提升,传统分析流程如ST Pipeline、zUMIs等面临计算效率瓶颈――处理单板3.6亿条reads需消耗128GB内存,耗时长达20小时。这种低效性严重制约了大规模药物筛选实验的开展。Merck & Co.的Scott Norton和John M. Gaspar团队在《Bioinformatics Advances》发表的DRAGoN(DRUG-seq/RNA-seq Analysis of Genetically-perturbed cells on Nextflow)研究,通过创新性的流程重构解决了这一难题。
关键技术方法包括:1)基于C++的预解复用模块,支持容错匹配(允许1个碱基错配);2)STAR 2.7.11b基因组比对与FeatureCounts 2.0.6基因定量组合;3)独创的UMI(Unique Molecular Identifier)纠错算法(10bp位置容差+1个编辑距离);4)Nextflow 24.10.6实现的分布式计算架构。测试数据来自Novartis的9个多孔板样本(GSE176150),每板含360个处理组。
研究结果显示:在计算性能方面,DRAGoN展现出显著优势。如图1b-d所示,其处理单板数据的中位运行时间(3.2小时)仅为ST Pipeline的1/6,峰值内存消耗(32GB)降低75%,同时保持UMI计数一致性(Pearson R2>0.95)。
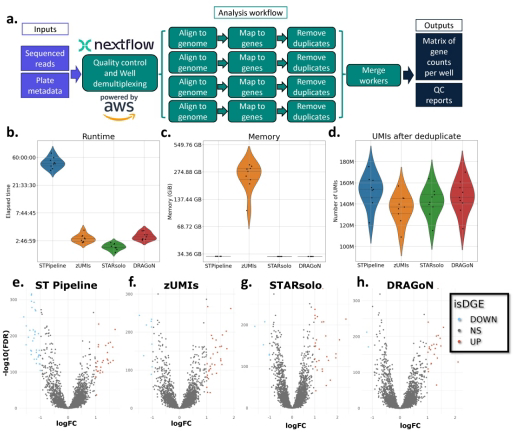
在生物学一致性方面,采用edgeR 3.4.0进行差异表达分析时,DRAGoN与STARsolo的log2FC相关性最高(R2=0.96),显著优于zUMIs(R2=0.89)。值得注意的是,在DMSO对照组间比较中,DRAGoN仅检测到48个假阳性差异基因,远低于STARsolo的153个,表明其具有更优的假阳性控制能力。
讨论部分强调,DRAGoN的创新性体现在两个维度:技术层面,前期解复用策略不仅实现计算资源优化,还支持按孔质量监控――例如通过UMI去重率异常可识别过度扩增的孔;应用层面,该流程兼容多种多映射reads分配策略(包括Uniform、EM算法等),为不同研究场景提供灵活性。作者指出,该工具已成功应用于Merck内部神经科学药物发现项目,其开源特性(MIT许可证)将促进更广泛的药物基因组学研究。
这项研究的里程碑意义在于:首次将工业级分布式计算理念引入DRUG-seq分析领域,使处理万级样本规模的药物筛选实验成为可能。正如作者在文末所述,随着单孔成本降至5美元以下,DRAGoN提供的分析效率将成为推动高通量转录组学在药物研发中规模化应用的关键引擎。
生物通微信公众号
知名企业招聘

