
-

生物通官微
陪你抓住生命科技
跳动的脉搏
生物通官微
陪你抓住生命科技
跳动的脉搏
迈向与CODIS STR谱系记录匹配的最小SNP集:优化法医遗传学中的反向兼容性
《European Journal of Human Genetics》:Toward minimal SNP sets for record-matching with CODIS STR profiles
【字体: 大 中 小 】 时间:2025年09月24日 来源:European Journal of Human Genetics 4.6
编辑推荐:
本研究针对法医遗传学中SNP与STR系统不兼容的难题,开发了一种基于连锁不平衡(LD)的记录匹配技术。通过系统筛选位于CODIS STRs附近1-Mb窗口内的SNP,研究人员发现仅需900个经过等位基因频率(MAF≥5%)和物理距离(≤0.125 Mb)双重筛选的SNP,即可在"大海捞针"场景中实现99%的匹配准确率。这项研究为开发反向兼容的法医SNP系统提供了关键理论依据,显著降低了多态性位点数量需求。
在法医遗传学领域,短串联重复序列(short tandem repeats, STRs)长期以来一直是个体识别的金标准。全球法医数据库已积累数百万个包含13-20个STR位点的基因档案。然而STR分析存在明显局限性:需要较高质量的DNA样本,对降解样本的检测能力有限,且不同实验室之间的位点组合可能存在差异。
与此同时,单核苷酸多态性(single nucleotide polymorphisms, SNPs)展现出独特优势。SNPs在基因组中分布密集,适合高通量分析,基因分型错误率低,尤其重要的是能够从高度降解的微量DNA样本中获取可靠数据。随着SNP分型技术的快速发展,开发新一代法医SNP系统的呼声日益高涨。但面临着一个关键挑战:如何实现新SNP系统与现有STR数据库的反向兼容?
传统的基因匹配要求使用相同的位点组合,而SNP和STR是完全不同的标记类型。为了解决这一难题,斯坦福大学的研究团队开发了基因记录匹配(genetic record-matching)技术,利用STRs与邻近SNPs之间的连锁不平衡(linkage disequilibrium, LD)来推断不同标记系统之间的关联性。早期研究表明,需要数万个随机选择的SNP才能实现准确匹配,但这对于实际应用来说仍然过多。
Tamara Gjorgjieva和Noah A. Rosenberg在《European Journal of Human Genetics》上发表的最新研究,致力于寻找能够实现高效记录匹配的最小SNP集合。研究人员使用1000 Genomes Project phase 3数据构建的参考面板,包含2504个来自全球26个人群样本的 phased SNP-STR单倍型数据。研究重点关注18个CODIS STR位点,并在每个STR周围1-Mb范围内筛选了192,672个SNP。
研究采用BEAGLE软件进行基因型插补,利用训练集(75%个体)的phased单倍型数据来推断测试集(25%个体)中隐藏的STR基因型。通过计算匹配得分矩阵,研究人员在四种匹配场景下评估准确性:一对一匹配、SNP查询、STR查询和大海捞针场景。最关键的是,他们比较了随机选择SNP与基于各种标准筛选的SNP的表现。
研究团队首先建立了基线性能:使用全部192,672个SNP时,在大海捞针场景中达到了0.998的中位匹配准确率。随机选择SNP需要至少9000个位点才能达到可比性能,这相当于约4.7%的基因组覆盖率。
然而当研究人员采用智能筛选策略时,结果令人振奋。基于最小等位基因频率(minor allele frequency, MAF)和群体MAF(pop-MAF)的筛选显著提高了效率:仅需1800个满足pop-MAF≥1%或pop-MAF≥5%条件的SNP就能达到全数据集性能。更令人惊讶的是,结合等位基因频率和物理距离的双重筛选策略进一步将需求降至900个SNP。
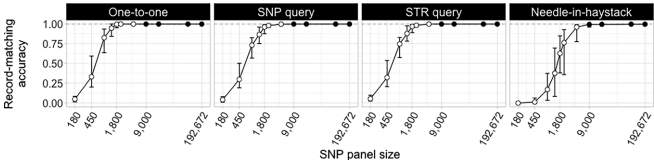
图1展示了随机选择SNP时的性能表现,可见随着SNP数量增加,匹配准确率逐步提高,但需要大量SNP才能达到理想效果。
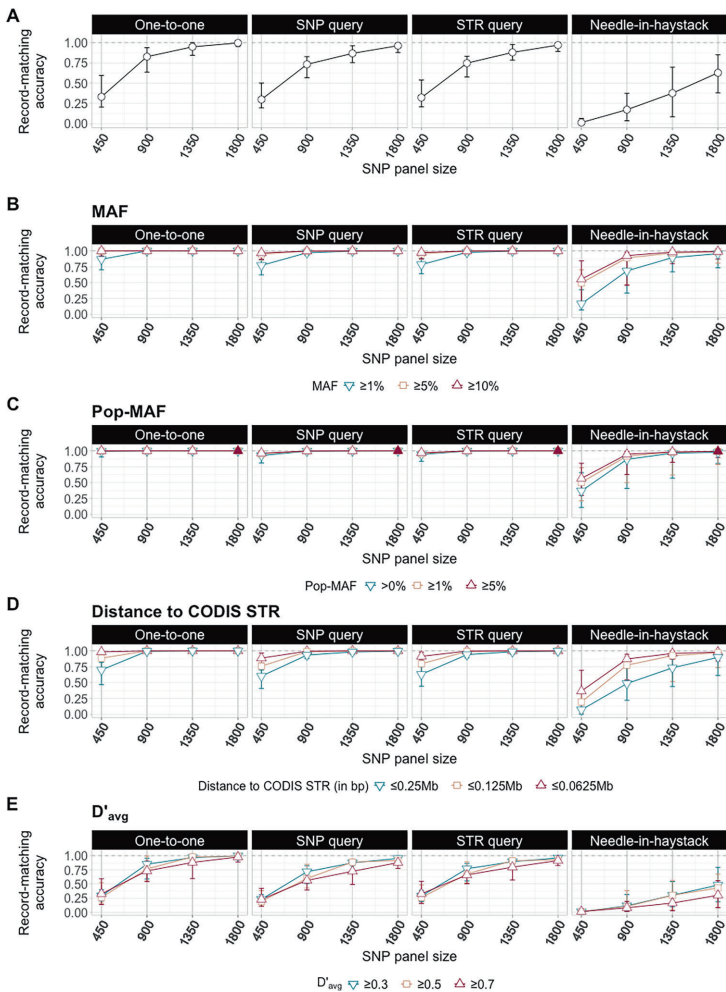
图2显示了基于不同特征筛选的SNP性能,其中MAF、pop-MAF和距离条件都显著优于随机选择,而D′avg条件表现相对较差。
最成功的组合包括:(MAF≥5%, 距离≤0.125 Mb)、(MAF≥10%, 距离≤0.125 Mb)、(pop-MAF>0%, 距离≤0.0625 Mb)、(pop-MAF>0%, 距离≤0.125 Mb)、(pop-MAF≥1%, 距离≤0.0625 Mb)和(pop-MAF≥5%, 距离≤0.125 Mb)。这些组合使用仅900个SNP就在大海捞针场景中实现了0.990-0.994的中位准确率。

图3进一步展示了双重筛选策略的优越性,多个条件组合都能以少量SNP实现高精度匹配。
值得注意的是,基于高强度LD(D′avg)的筛选策略效果不佳,研究人员推测这可能是因为高LD的SNP之间存在冗余信息。相反,等位基因频率筛选确保了训练集中有足够的次要等位基因拷贝,使BEAGLE能够准确学习SNP与多态性STR之间的相关性。
研究还发现,记录匹配准确率在不同超级群体间存在差异,从高到低依次为:非洲(AFR)、美洲(AMR)、东亚(EAS)、欧洲(EUR)和南亚(SAS)。使用pop-MAF条件有助于确保所选SNP在多个群体中都具有多态性,从而支持跨群体的记录匹配。
研究人员通过推估发现,在精心选择的900个SNP集中,约有70%的真实匹配可以在大群体中达到109的后验先验 odds 比,表明这些SNP集在真实法医场景中具有实用价值。
这项研究为开发下一代法医SNP系统提供了重要启示。一个理想的法医SNP面板可以包含多个功能类别的SNP:用于个体识别的核心SNP、用于祖先推断的SNP,以及用于实现与现有CODIS STR数据库反向兼容的记录匹配SNP。研究表明,只需不到1000个精心选择的SNP就能支持后一种功能。
从隐私保护角度,较小的SNP集合也降低了意外表型预测的风险,这符合法医遗传学中"表型隐私"的设计原则。然而研究人员也强调,记录匹配技术本身涉及隐私权衡,因为它使得不同数据库之间的身份链接成为可能。
这项研究展示了记录匹配技术在小型SNP面板中的巨大潜力,为最终用SNP系统替代STR系统铺平了道路。未来的研究需要在更大样本中验证这些发现,并开发组合优化算法来进一步减少所需SNP数量,同时评估各种性能指标以确保实际应用的可靠性。
生物通微信公众号

