
-

����ͨ��
����ץס�����Ƽ�
����������
����ͨ��
����ץס�����Ƽ�
����������
ţ�����׳����������ڻ����������ѧ�е��Զ���Ӧ�ã�����Ч���������ԵĹؼ��о�
�����壺 �� �� С �� ʱ�䣺2025��09��27�� ��Դ��BMC Genomics 3.7
�༭�Ƽ���
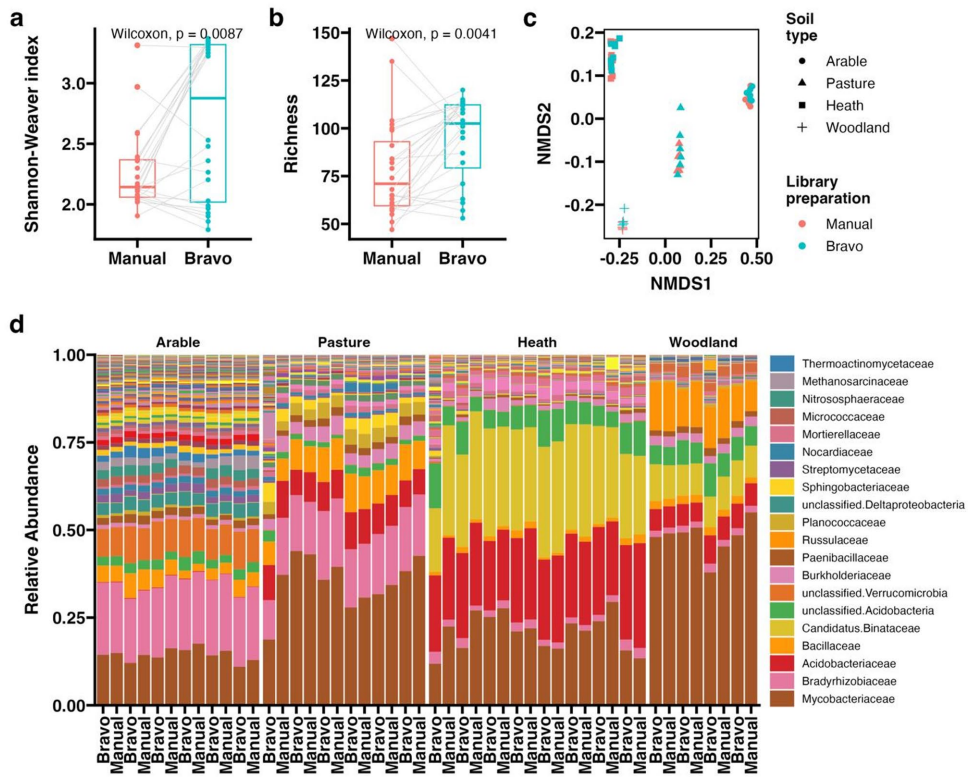
�������о���Ի����������ѧ�г���������ONT���Զ����������̵���֤����ͨ���Ա��ֶ����Զ�����Bravoƽ̨��������֤ʵ�Զ��������ڱ�������Ⱥ��ṹһ���Ե�ͬʱ��������ϡ�����ּ���ʺͷ���Ч�ʣ�Ϊ��ͨ�����������Ժ�������о��ṩ�˹ؼ�����֧�֡�

 ����ͨ�Ź��ں�
����ͨ�Ź��ں�
֪����ҵ��Ƹ

