《Cellular Oncology》:The histone acylation network: emerging therapeutic targets for remodeling the epigenetic landscape in pancreatic ductal adenocarcinoma
编辑推荐:
本文深入探讨了组蛋白酰化修饰网络在胰腺导管腺癌(PDAC)中的核心调控作用。作者系统综述了从经典的乙酰化(Kac),到与肿瘤代谢重编程紧密相连的新型修饰如乳酸化(Kla)、琥珀酰化(Ksucc)和丙酰化(Kpr),阐明了它们如何通过共享的“书写器”(如p300)和“擦除器”(如HDACs)形成动态、复杂的调控网络,协同或拮抗地调控MYC、GATA6等关键基因,驱动PDAC的增殖、转移、免疫逃逸及治疗抵抗。文章不仅揭示了这些修饰作为肿瘤发生新驱动因子的潜力,还评估了靶向该网络的表观遗传药物临床前景,为克服当前治疗困境提供了新思路。
胰腺导管腺癌(PDAC)是一种致死率极高的消化系统恶性肿瘤,其复杂的分子调控网络是导致预后不佳的主要原因。在众多调控机制中,表观遗传修饰,特别是组蛋白翻译后修饰(PTMs),扮演着关键角色。组蛋白酰化网络,作为一个核心的调控枢纽,通过动态重塑染色质景观,深刻影响着PDAC的生物学行为。这篇综述旨在系统阐述多样化的组蛋白酰化修饰在PDAC中的功能、机制及相互作用网络,并展望其作为新兴治疗靶点的巨大潜力。
组蛋白酰化的分子基础与调控逻辑
不断扩展的组蛋白酰化修饰库
超越经典的组蛋白乙酰化(Kac),质谱技术的进步揭示了多种短链酰化修饰的存在,包括丙酰化(Kpr)、丁酰化(Kbu)、琥珀酰化(Ksucc)、巴豆酰化(Kcr)、丙二酰化(Kma)、2-羟基异丁酰化(Khib)、β-羟基丁酰化(Kbhb)以及乳酸化(Kla)。这些修饰作为分子桥梁,直接将代谢中间产物与表观遗传调控联系起来。
化学特性与功能启示
这些酰化修饰可根据其化学性质分为三大类:
- 1.
疏水性酰基(如Kpr、Kbu、Kcr):增加空间位阻和疏水性。例如,Kcr的刚性平面π-键可特异性增强与YEATS结构域蛋白(如AF9)的亲和力。
- 2.
极性酰基(如Kbhb、Khib、Kla):带有羟基,便于形成氢键,有助于招募染色质重塑复合物。Kla具有独特的动力学特征,积累缓慢,可编码持续的代谢信号。
- 3.
酸性修饰(如Kma、Ksucc、Kglu):诱导电荷从正性反转为负性,类似于磷酸化,深刻影响染色质结构。
酰化调控的酶学与代谢机制
组蛋白酰化是一个动态可逆的过程。
- •
共享的酶促机器:许多非经典酰化与乙酰化共享经典的“书写器”和“擦除器”系统,如p300和组蛋白去乙酰化酶(HDACs)。p300因其底物结合口袋的结构灵活性,可作为“代谢传感器”,根据波动酰基辅酶A(Acyl-CoA)池的比例切换其底物偏好。
- •
代谢底物供应:修饰过程依赖于相应的酰基辅酶A供体(如乙酰辅酶A、乳酸辅酶A)。在PDAC等高糖酵解背景下,乳酸的积累会引导p300优先催化组蛋白乳酸化。
- •
特异性酶:同时也存在特异性酶,如乳酸化修饰的特异性“书写器”AARS1/2。
PDAC中组蛋白酰化概述
组蛋白酰化在PDAC中充当中心调控枢纽,影响包括代谢重编程、上皮-间质转化(EMT)、增殖存活、免疫逃逸、肿瘤微环境(TME)互作以及细胞命运(分化与干性)在内的多个关键生物学过程。
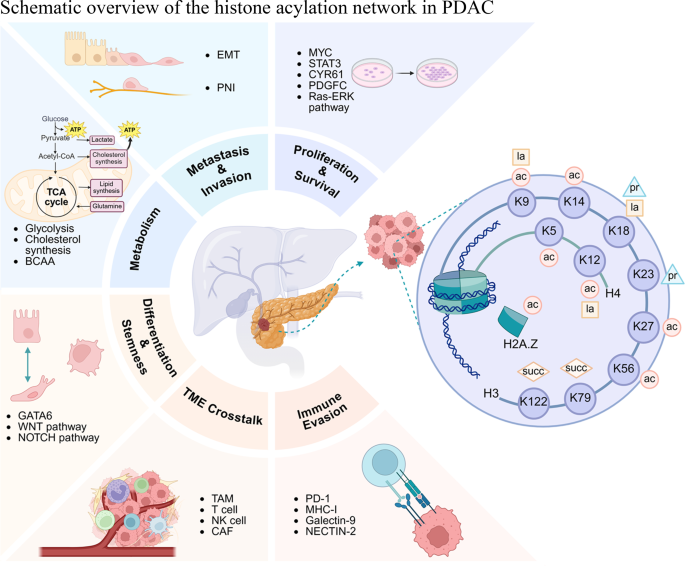
组蛋白乙酰化在PDAC中的功能与机制
组蛋白乙酰化是研究最深入的修饰,在PDAC的细胞分化、增殖转移、凋亡、免疫逃逸及TME互作中均发挥关键作用。
- •
细胞分化与亚型:H3K27ac是PDAC表型分化的关键调节因子。p300通过直接调控GATA6的转录维持经典亚型分化。而DNA去甲基化酶TET3可招募HDACs至GATA6启动子区,导致局部H3K27去乙酰化和GATA6转录抑制,从而促进去分化的基底样亚型转变。
- •
增殖与转移: Ras-ERK1/2信号通路通过介导组蛋白乙酰转移酶PCAF的降解,导致全基因组H3K9ac水平降低,从而激活CYR61、WNT16B等促癌基因转录。RARG可增强MYC、STAT3、SLC2A1等基因启动子区的H3K27ac,促进肿瘤生长。组蛋白伴侣ASF1B与CBP合作,特异性促进MYC启动子区的H3K56ac以激活其转录。此外,p300还能通过增强PDGFC启动子区的H3K27ac来上调其表达,促进肿瘤侵袭。
- •
免疫逃逸: 组蛋白乙酰转移酶1(HAT1)通过乙酰化H4K5和H4K12,被BRD4识别,进而上调PD-L1转录。HDAC抑制剂如丙戊酸(VPA)可通过增加全局组蛋白乙酰化和直接乙酰化PD-L1蛋白来上调其表达。另一方面,泛HDAC抑制剂LAQ824可通过增加抗原呈递基因(如MHC-I)启动子区的H3K27ac水平,上调MHC-I表达,抑制免疫逃逸。
- •
肿瘤微环境: 癌症相关成纤维细胞(CAFs)来源的外泌体(CAF-exos)中的miR-421可抑制PDAC细胞中的SIRT3,增加H3K9ac,激活HIF-1α,促进糖酵解和肿瘤生长。在CAFs中,p300与GLI1形成的复合物以依赖于H4ac/H3K27ac/H3K14ac的方式增强SDF1表达,促进癌细胞迁移。反之,PDAC细胞中SETD2的缺失导致Bmp2启动子区异常的H3K27ac积累,增强其表达。分泌的BMP2可诱导邻近的成纤维细胞分化为一种独特的脂质丰富型CAF亚型,为癌细胞提供代谢支持。
组蛋白乳酸化在PDAC中的功能与机制
在高度糖酵解的PDAC中,组蛋白乳酸化(Kla)作为代谢-表观遗传轴的核心,显著影响肿瘤的恶性特征。
- •
肿瘤进展调控:糖酵解来源的乳酸可提高MESP1启动子区的H3K18la水平,驱动EMT、增强增殖迁移并抑制凋亡。CaMKIIδ-SIRT4-ENO1信号轴通过驱动糖酵解依赖的组蛋白乳酸化,维持肿瘤起始细胞(TICs)的干性。H3K18la还能富集于TTK和BUB1B等有丝分裂调节因子启动子区,驱动细胞增殖,并通过p300上调和TTK介导的LDHA激活形成正反馈环路。高血糖可抑制AMPK活性,激活DRP1介导的线粒体分裂和乳酸生成,进而提高H3K18la水平,再激活TTK/BUB1B信号。另一个反馈环路涉及H3K18la在ACAT2启动子区的富集,ACAT2通过乙酰化稳定MTCH2,损害线粒体氧化磷酸化(OXPHOS),迫使细胞更依赖糖酵解,从而增加乳酸和H3K18la水平。
- •
肿瘤微环境重塑: 乳酸-乳酸化轴是TME重塑的主要驱动力。上述ACAT2-MTCH2反馈环路上调胆固醇合成,并通过小细胞外囊泡(sEVs)转移胆固醇,将肿瘤相关巨噬细胞(TAMs)重编程为M2表型,促进免疫逃逸。CTCF/HNRNPU/FLG-AS1复合物可招募p300催化IGF2BP2启动子区的H3K18la,上调IGF2BP2稳定CSF1 mRNA,增加CSF1分泌,进而激活TAMs上的CSF1R,诱导免疫抑制性M2极化。H4K12la可增强染色质可及性,激活NECTIN2、LGALS9等免疫检查点基因,抑制T/NK细胞功能。此外,糖酵解的神经侵犯相关CAFs(pCAFs)分泌乳酸至TME,提高PDAC细胞中的H3K18la水平,激活L1CAM、SLIT1等神经侵袭相关基因,促进神经周围侵犯(PNI)。
其他组蛋白酰化在PDAC中的功能与机制
- •
组蛋白丙酰化: 支链氨基酸(BCAA)代谢,特别是异亮氨酸(Ile)分解,是PDAC细胞中丙酰辅酶A(pr-CoA)和组蛋白丙酰化(如H3K23pr、H3K18pr)的主要来源。完整的Ile分解代谢通路可发生非经典核转位,在细胞核内直接为组蛋白修饰提供底物。该修饰主要由KAT7书写,由HDAC3擦除。
- •
组蛋白琥珀酰化: 琥珀酰化是癌症细胞代谢的关键调节机制。KAT2A介导的H3K79succ可上调YWHAZ/14-3-3ζ表达,从而稳定β-连环蛋白(β-catenin),促进糖酵解、增殖、迁移侵袭和EMT。HAT1可增加CBP、BPTF、RPTOR等基因启动子区的H3K122succ水平,并能催化糖酵解酶PGAM1的K99琥珀酰化,增强其酶活,促进糖酵解通量。
- •
组蛋白巴豆酰化与2-羟基异丁酰化: 在PDAC中,巴豆酰化广泛修饰代谢通路相关酶。2-羟基异丁酰化(Khib)可能通过调节糖酵解促进癌症进展,p300可特异性介导糖酵解关键酶上的Khib修饰而不影响其乙酰化。抑制Khib转移酶KAT5可降低Khib水平并抑制PDAC细胞增殖、迁移和侵袭。
PDAC中组蛋白酰化的交互对话
组蛋白酰化在PDAC中作为一个高度整合的网络运行,涉及共享的酶促机器,并能将不同的代谢信号汇聚到关键的基因组位点,通过广泛的表观遗传交互对话决定细胞命运。
内部基因调控的整合: 关键癌基因和谱系因子的强劲表达由多种酰化修饰汇聚确保。例如,MYC的强表达由H3K27ac、H3K56ac和H3K79succ的汇聚所保证,这些标记通过不同的互补通路沉积。相反,谱系因子GATA6受p300介导的H3K27ac(维持分化)与TET3招募HDACs(擦除此标记以驱动向基底样亚型转变)之间的动态竞争控制。
肿瘤微环境的交互对话: 酰化网络是协调肿瘤细胞与其微环境关系的桥梁。组蛋白乳酸化作为糖酵解通量的主要传感器,驱动与TAMs的交互对话。同样,该网络协调了与CAFs的互惠对话,涉及外泌体miR-421和BMP2信号交换,以调节H3K9ac和H3K27ac水平。这些复杂的相互作用共同塑造了免疫抑制和促肿瘤的TME。
总结与展望
对失调的组蛋白酰化网络的全面解读,不仅揭示了胰腺肿瘤发生的新致病驱动因子,也为精准肿瘤学建立了机制基础。靶向该网络的表观遗传药物,或与常规/靶向疗法联用,展现出克服当前治疗局限性和治疗抵抗的潜力。未来的研究需要进一步阐明不同酰化修饰之间复杂的交互对话,开发更具选择性的抑制剂,并将这些基础研究发现转化为临床有效的治疗策略。




